


基于CDE、FDA、EMA 2025年度数据的效率评估与趋势研判
数据来源:CDE 2025年度药品审评报告、FDA CDER年报、EMA Human Medicines Highlight
目录
一、报告摘要与核心发现
1.1 研究背景与目的
1.2 核心发现概要
二、2025年度三大监管机构审评数据概览
2.1 中国NMPA/CDE审评数据
2.2 美国FDA审评数据
2.3 欧洲EMA审评数据
2.4 关键指标横向对比
三、审评效率多维度比较分析
3.1 批准数量趋势对比(2020-2025)
3.2 药品类型与结构差异
3.3 治疗领域分布特征
3.4 加快审评程序机制比较
四、中国创新医药发展趋势深度分析
4.1 创新药批准数量持续攀升
4.2 国产创新药占比显著提升
4.3 新兴治疗领域布局加速
4.4 监管科学与国际化进程
五、结论与期待
5.1 主要结论
5.2 期待
参考文献
一、报告摘要与核心发现
1.1 研究背景与目的
药品审评审批是保障公众用药安全、促进医药产业创新的核心环节。随着全球医药产业的快速发展和创新药物研发的不断突破,各国药品监管机构在审评效率、审批标准、创新激励等方面呈现出不同的特点和趋势。中国药品审评中心(CDE)近年来持续推进审评审批制度改革,在提高审评效率、鼓励药物创新方面取得了显著成效。本报告基于CDE发布的《2025年度药品审评报告》,结合美国食品药品监督管理局(FDA)和欧洲药品管理局(EMA)同期的审评数据,对三大监管机构的审评效率与区别进行系统比较,并深入分析中国创新医药的发展趋势。
本研究的数据来源主要包括三个方面:CDE 2025年度药品审评报告(涵盖受理量、审结量、创新药批准、加快上市程序使用等详细数据)、FDA药物评价与研究中心(CDER)及生物制品评价与研究中心(CBER)发布的年度新药批准数据,以及EMA发布的人类用药年度亮点报告(Human Medicines Highlights)。通过对这些权威数据的系统整理和比较分析,旨在揭示三大监管机构的审评特征差异,评估中国药品审评体系的改革成效,并为未来政策优化提供参考依据。
1.2 核心发现概要
发现一:中国创新药批准量持续领先
2025年,NMPA共批准1类创新药76个品种,创历史新高,连续第二年超过FDA(46个CDER NMEs)和EMA(38个NAS)。2019年至2025年,NMPA创新药批准的复合增长率高达30.6%,远超FDA的-2.8%和EMA的-0.5%。
发现二:审评效率差距显著缩小
CDE通过突破性治疗药物程序、附条件批准、优先审评审批等多种加快上市程序,大幅缩短了创新药审评时间。2025年,CDE纳入突破性治疗药物程序101项,附条件批准38项,优先审评审批124项,各项数据均呈上升趋势。同时,30日临床试验审评通道的开通进一步优化了审评效率。
发现三:中国创新药结构持续优化
2025年批准的76个创新药中,化学药品46个、生物制品25个、中药5个,涵盖抗肿瘤、抗感染、内分泌代谢等多个治疗领域。全年批准罕见病用药48个品种、儿童用药138个品种,充分体现了以临床需求为导向的审评理念。
发现四:监管国际化进程加速推进
CDE深度参与ICH国际协调,2025年当选ICH MedDRA指导委员会成员,累计采纳实施73个ICH指导原则。同时,中国药品监管科学研究蓬勃开展,在细胞与基因治疗、创新疫苗、放射性药物等前沿领域建立了系统的技术评价体系。
二、2025年度三大监管机构审评数据概览
2.1 中国NMPA/CDE审评数据
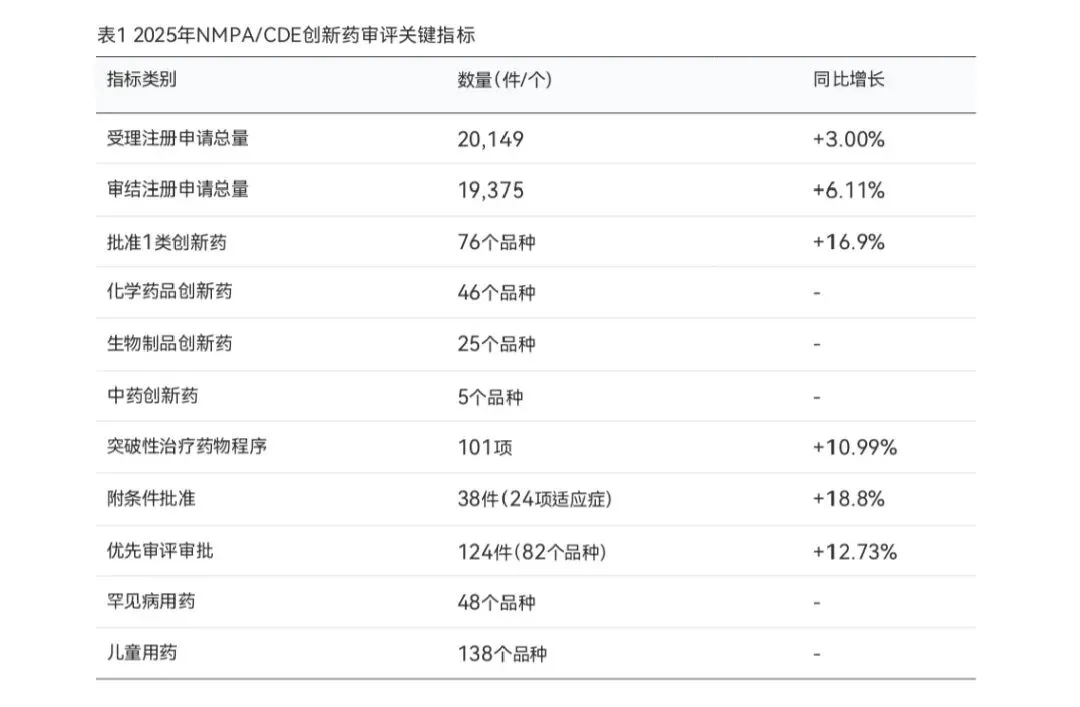
根据CDE发布的2025年度药品审评报告,全年共受理各类注册申请20,149件(以受理号计,同比增加3.00%),审结各类药品注册申请19,375件(同比增加6.11%)。其中,技术审评类注册申请受理16,130件,包括中药注册申请2,723件、化学药品注册申请10,587件、生物制品注册申请2,820件。这些数据反映出中国药品注册申请量保持稳步增长态势,药审中心的审评能力持续提升。
在创新药审评方面,2025年NMPA共批准1类创新药76个品种,其中化学药品46个、生物制品25个、中药5个。这一数字较2024年的65个增长了16.9%,再创历史新高。从批准结构来看,化学药品创新药占比约60.5%,生物制品创新药占比约32.9%,中药创新药占比约6.6%。全年通过优先审评审批程序批准药品124件(82个品种),通过附条件批准程序批准药品38件(24项适应症),纳入突破性治疗药物程序101项(89个适应症)。
值得关注的是,CDE在特殊人群用药审评方面表现突出。2025年批准罕见病用药48个品种,其中12个品种(24.5%)通过优先审评审批程序加快上市;批准儿童用药138个品种(含98个上市许可申请),其中25个品种(18.1%)通过优先审评审批程序加快上市,另有40个品种扩展儿童适应症。此外,全年批准放射性药品6个品种,其中3个品种通过优先审评审批程序加快上市,反映出CDE在满足临床急需用药方面的积极努力。
2.2美国FDA审评数据
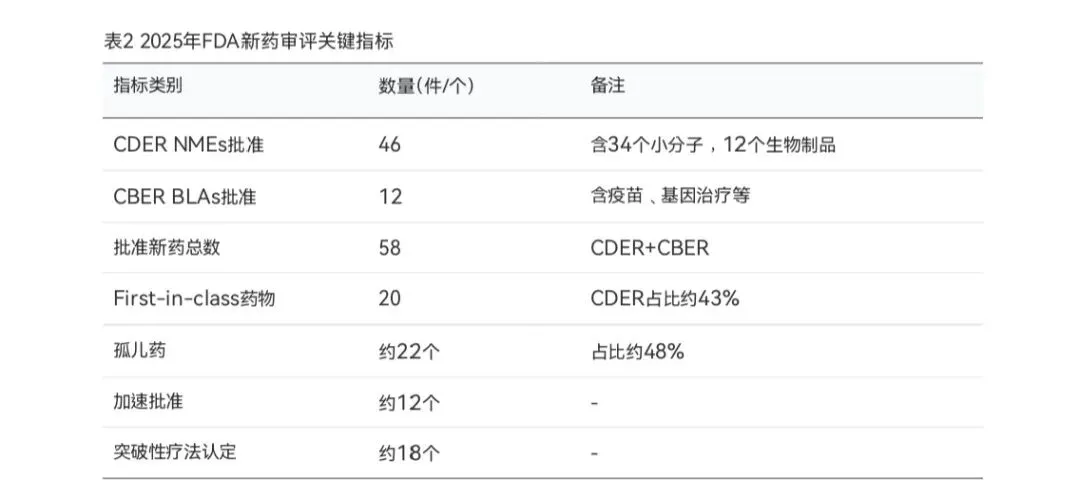
2025年,FDA药品审评工作面临多重挑战。根据FDA药物评价与研究中心(CDER)发布的数据,全年共批准46个新分子实体(NMEs),较2024年的50个和2023年的55个有所下降,但仍高于历史平均水平。此外, 生物制品评价与研究中心( CBER) 批准了12个生物制品许可申请
(BLAs),包括疫苗、基因治疗等创新产品,使得FDA 2025年批准的新药总数达到58个。
从药品类型分析,CDER批准的46个NMEs中,小分子药物34个,生物制品12个(包括单克隆 抗体、抗体药物偶联物ADC、融合蛋白等)。值得关注的是,2025年有2个ADC药物获批,结束了 此前连续两年无ADC批准的局面。生物制品占CDER批准总数的26%,略低于2018-2024年间29% 的平均水平。在CBER方面,批准的12个BLAs包括2个新冠疫苗、基孔肯雅疫苗、脑膜炎球菌疫苗 等。
在治疗领域方面,肿瘤依然是FDA批准新药的首要领域。2025年批准的46个NMEs中,约39%用于肿瘤适应症。此外,神经系统疾病、内分泌代谢疾病、罕见病等领域的创新药也占据了
相当比例。FDA在2025年批准了多个具有里程碑意义的药物,包括首个治疗脑腱黄瘤病的药物Ctexli、首个获批用于成人和儿童1型神经纤维瘤病相关丛状神经纤维瘤的MEK抑制剂Gomekli 等。
2.3欧洲EMA审评数据
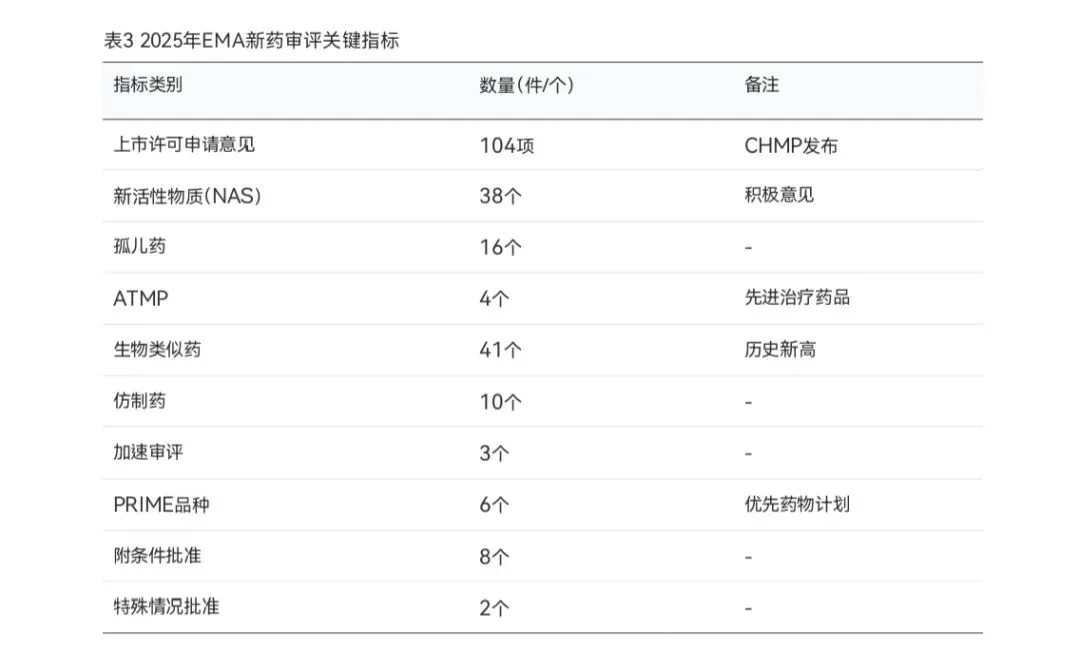
2025年,欧洲药品管理局(EMA)的人类用药委员会(CHMP)共发布了104项关于人用药 品上市许可申请的意见,其中包括38个新活性物质(NAS)的积极意见。全年批准的孤儿药16 个、先进治疗药品(ATMP)4个、生物类似药41个(创历史新高),另有10个仿制药获得推荐。此外,CHMP发布了7项消极意见,22项申请在审评过程中由申请人撤回。
EMA在2025年特别强调了多项具有重大临床价值的创新药批准。在肿瘤与细胞治疗领域, Anktiva(用于高风险非肌层浸润性膀胱癌)获得附条件批准,Aucatzyl(CAR-T细胞治疗复发 难治B细胞急性淋巴细胞白血病,属ATMP和PRIME品种)和Zemcelpro(脐带血干细胞治疗, 属ATMP和孤儿药)获得积极意见。在罕见病与遗传病领域,Vyjuvek(营养不良性大疱性表皮松 解症基因治疗)和Waskyra(Wiskott-Aldrich综合征首个基因治疗)等药物获得批准,展现了EMA在鼓励罕见病治疗创新方面的持续努力。
生物类似药是2025年EMA审评工作的一大亮点。全年批准的41个生物类似药创下历史新高, 其中23个参照药为地舒单抗。自2005年建立生物类似药监管框架以来,EMA已累计审评181项申 请,批准165个生物类似药。相比之下,FDA累计批准的生物类似药约为81个,充分显示了EMA 在生物类似药审评领域的全球领先地位。
2.4 关键指标横向对比
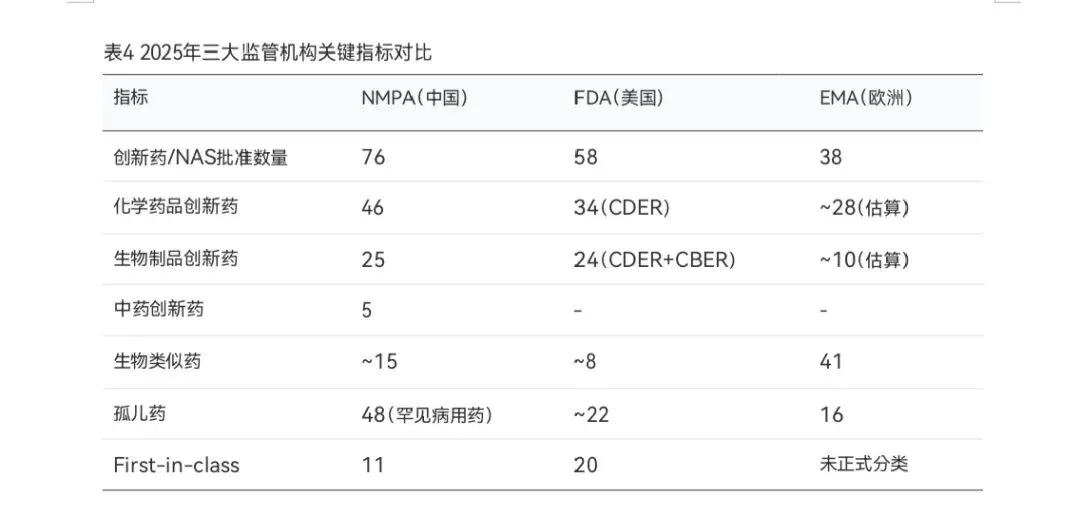
综合对比三大监管机构2025年的审评数据,可以清晰地观察到各自的特点和差异。在创新药批准数量方面,NMPA以76个1类创新药位居首位,FDA以58个新药(CDER+CBER)紧随其后,EMA以38个新活性物质排名第三。需要注意的是,由于各机构的药品分类标准和统计口径存在差异,直接数量比较仅供参考。
从药品结构来看,NMPA的批准数据包含化学药品、生物制品和中药三大类,体现了中国医药产业的多元化特征。其中中药创新药5个是中国特有的药品类别,反映了CDE在传承与创新并重的审评理念。FDA和EMA的批准数据则主要集中在化学药品和生物制品领域。在生物类似药方面,EMA以41个批准量遥遥领先,反映了欧洲对生物类似药的积极接纳态度。
三、审评效率多维度比较分析
3.1 批准数量趋势对比(2020-2025)
通过对2020年至2025年三大监管机构新药批准数量的纵向比较,可以揭示出各自的发展趋势 和增长动力。FDA在2020年至2025年间,新药批准数量在37-55个之间波动,5年间总体呈现轻微 下降趋势,年均复合增长率为-2.8%。EMA同期的NAS批准数量在32-42个之间波动,年均复合增 长率约为-0.5%,基本保持稳定。相比之下,NMPA的创新药批准数量从2020年的20个增长至2025年的76个,年均复合增长率高达30.6%,增长势头最为强劲。
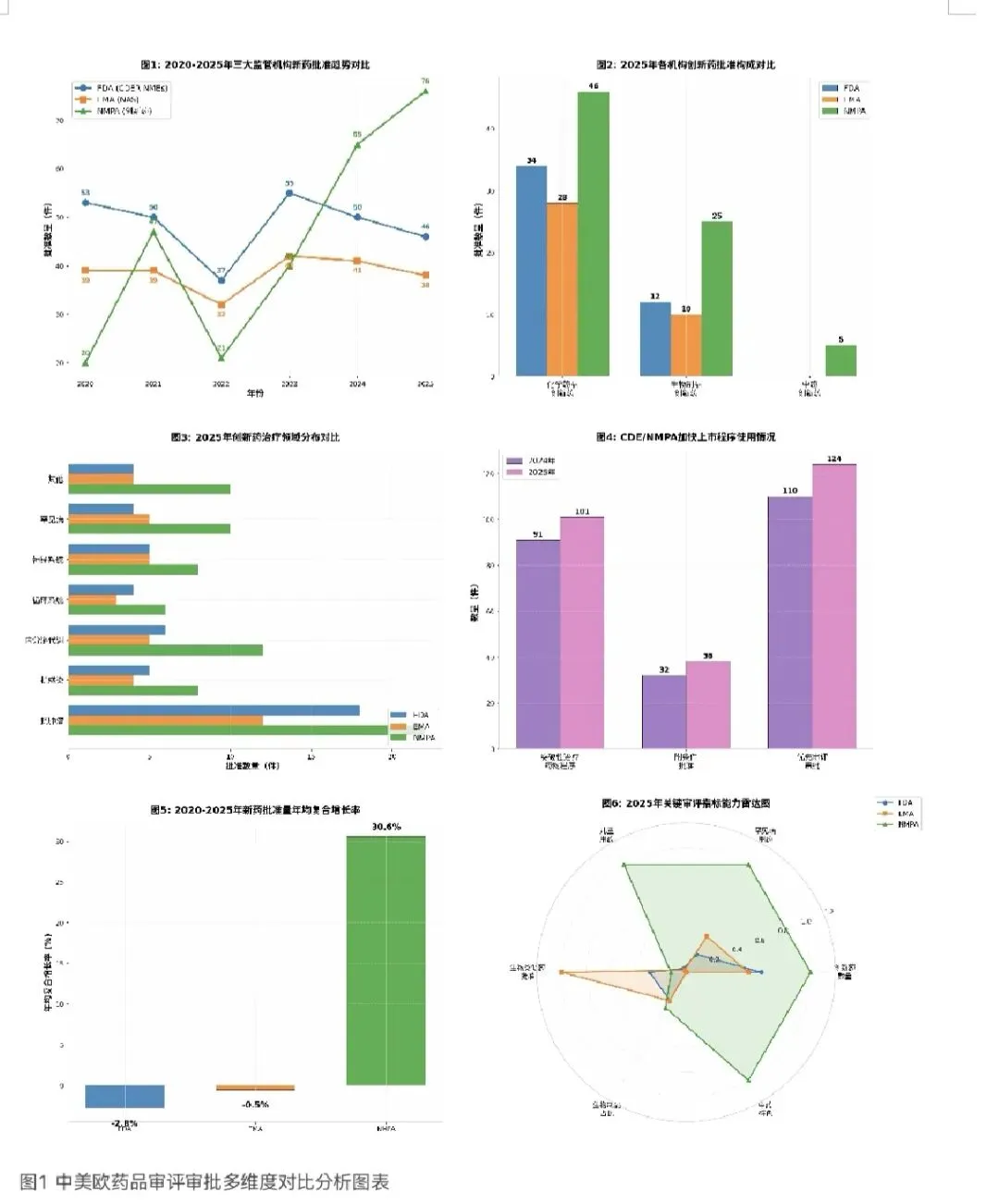
从趋势演变来看,NMPA创新药批准量在2020-2021年间经历了第一次跃升(从20个增至47 个),随后在2022年有所回落至21个,但从2023年开始进入持续快速增长阶段,2023年40个,2024年65个、2025年76个,三年间增长了90%。这一增长趋势与CDE持续推进的审评审批制度改革密切相关,包括优先审评审批、突破性治疗药物程序、附条件批准等多种加快上市程序的建立和完善,显著提升了审评效率和创新药可及性。
FDA的批准趋势则显示出一定的波动性,2022年受新冠疫情影响批准量降至37个,2023年反 弹至55个,但2024年和2025年连续两年下降。这种波动可能与FDA的政治环境变化、人员变动等 系统性因素有关,而非制药行业本身创新能力的下降。EMA的批准数量在过去五年间相对稳定, 保持在32-42个的区间内,体现了欧洲药品审评体系的成熟和稳定。
3.2 药品类型与结构差异
三大监管机构在药品类型和结构方面存在显著差异,这反映了各自医药产业的特点和监管重点。NMPA的批准结构最为多元,涵盖化学药品、生物制品和中药三大类。2025年批准的76个创新药中,化学药品46个(60.5%),生物制品25个(32.9%),中药5个(6.6%)。这一结构与中国的医药产业格局相符,化学药品仍然是创新主力,但生物制品的占比正在逐年提升,反映了行业向生物药转型的趋势。
FDA的药品结构以化学药品为主导,2025年CDER批准的46个NMEs中化学药品34个(73.9%),生物制品12个(26.1%)。但如果将CBER批准的12个BLAs纳入统计,则生物制品总数达到24个,占FDA批准新药总数的41.4%,显示出FDA在生物制品审评方面的强大能力。FDA在2025年批准的生物制品包括9个单克隆抗体、2个ADC和1个融合蛋白,其中ADC领域在连续两年空白后重新获得突破。
EMA在生物类似药审评方面表现突出,2025年批准的41个生物类似药创下历史新高,累计批准量达到165个,远超FDA的81个。这一成就得益于EMA自2005年建立的成熟生物类似药监管框架和科学的审评标准。在创新药方面,EMA批准了4个ATMP,包括CAR-T细胞治疗和基因治疗产品,在先进治疗领域保持了领先地位。
3.3 治疗领域分布特征
在治疗领域分布方面,三大监管机构均将肿瘤作为创新药审评的首要领域。NMPA 2025年批准的化学药品和生物制品创新药中,抗肿瘤药物占比约37%,是最大的治疗领域。这一比例与FDA的情况相似,FDA 2025年批准的新药中约39%用于肿瘤适应症。EMA在肿瘤领域同样表现活跃,批准了多个重要的肿瘤免疫治疗药物和ADC产品。
除肿瘤外,各机构的治疗领域分布也体现出不同的特点。NMPA在抗感染领域表现突出,2025年批准了多个国产抗流感新药(如昂拉地韦、玛帕西沙韦、玛舒拉沙韦、玛硒洛沙韦等)和抗新冠药物,反映了CDE在应对公共卫生事件方面的快速响应能力。在内分泌代谢领域,NMPA批准了多个GLP-1类药物和糖尿病治疗新药,与全球医药研发趋势保持一致。
FDA在罕见病领域继续保持领先地位,2025年批准的46个NMEs中约48%获得了孤儿药认定。FDA还批准了多个具有里程碑意义的罕见病治疗药物,如首个治疗脑腱黄瘤病的Ctexli。EMA在罕见病领域同样表现积极,2025年批准的38个NAS中有16个孤儿药(42.1%),并批准了多个ATMP用于罕见遗传病治疗。
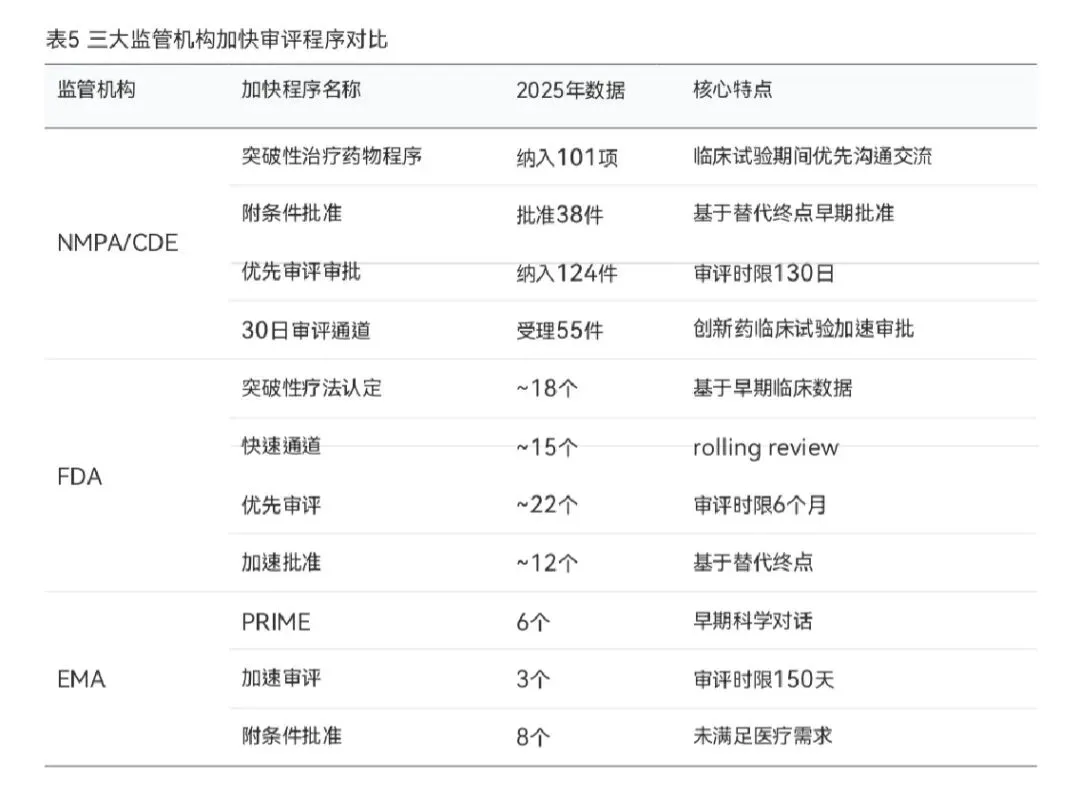
3.4 加快审评程序机制比较
三大监管机构均建立了较为完善的加快审评程序体系,但在具体机制和命名上存在差异。NMPA/CDE建立了"突破性治疗药物程序-附条件批准-优先审评审批"三位一体的加快上市注册程序体系。2025年,CDE收到突破性治疗药物程序申请424件,同意纳入101件(23.82%),较2024年增长10.99%;纳入优先审评审批133件(92个品种),同比增长7.26%;附条件批准38件(24项适应症)。此外,CDE在2025年9月推出30日临床试验审评通道,截至年底已受理55件、完成审评20件,进一步优化了创新药的临床开发效率。
FDA的加快审评程序包括突破性疗法认定(Breakthrough Therapy)、快速通道(Fast Track)、优先审评(Priority Review)和加速批准(Accelerated Approval)四种。2025年,FDA在46个NMEs中授予约18个突破性疗法认定,约22个优先审评,约12个加速批准。FDA的孤儿药认定数量继续保持高水平,约22个新药获得孤儿药认定。
EMA的加快审评程序包括优先药物计划(PRIME)、加速审评(Accelerated Assessment)和附条件批准(Conditional Marketing Authorisation)。2025年,6个品种获得PRIME资格, 3个品种通过加速审评程序,8个品种获得附条件批准,2个品种在特殊情况下获得授权。此外, EMA在2025年发布了7项消极意见,体现了其在获益-风险评估方面的审慎态度。
四、中国创新医药发展趋势深度分析
4.1 创新药批准数量持续攀升
中国创新药批准数量自2019年以来呈现出持续快速增长的态势,年均复合增长率达到30.6%,远高于全球其他主要市场。这一增长趋势的背后,是CDE持续推进的审评审批制度改革和创新驱动发展战略的深入实施。从数据演变来看,NMPA批准的创新药数量从2020年的20个增至2025年的76个,五年间增长了280%。
关键数据:2025年批准的76个创新药中,26个品种(34.2%)通过优先审评审批程序批准上市,15个品种(19.7%)附条件批准上市,15个品种(19.7%)在临床试验期间纳入了突破性治疗药物程序。这些数字充分表明,加快上市程序已成为推动创新药快速上市的重要机制。
从年度增长趋势分析,2023年是重要的转折点。当年创新药批准量达到40个,较2022年的21个几乎翻倍,标志着CDE审评能力质的飞跃。2024年进一步增长至65个,2025年达到76个,增速虽然有所放缓但基数已经大幅提高。这一增长趋势与中国医药产业研发投入的持续增长、创新能力的不断提升以及审评体系的持续优化密切相关。
值得关注的是,CDE在2025年实现了"双创新高":不仅创新药批准总数76个创历史新高,新机制新靶点药物11个也创历史新高。这表明中国创新药审评不仅追求数量增长,更注重质量和创新性。全年批准的11个新机制新靶点药物涵盖first-in-class和best-in-class品种,体现了中国创新药研发正在从跟随式创新向源头创新转型。
4.2 国产创新药占比显著提升
2025年批准的76个创新药中,国产创新药占比持续提升,反映出中国医药产业自主创新能力的显著增强。从药品注册分类来看,化学药品1类创新药和生物制品1类创新药中,本土企业研发的品种占据了主导地位。以抗肿瘤领域为例,全年批准的多个国产创新药涵盖了靶向治疗、免疫治疗等多个细分方向,包括多款针对KRAS G12C突变、EGFR突变、HER2阳性等热门靶点的创新药物。
国产创新药的崛起在多个治疗领域均有体现。在抗感染领域,CDE在2025年批准了多款国产抗流感新药,包括昂拉地韦、玛帕西沙韦、玛舒拉沙韦、玛硒洛沙韦等,这些药物的上市为应对季节性流感提供了更多治疗选择。在内分泌代谢领域,信达生物的玛仕度肽注射液和派格生物的维培那肽注射液等GLP-1类创新药的批准,标志着中国企业在代谢性疾病治疗领域取得重要突破。
国产创新药的快速发展得益于多方面的因素。首先,国家药品监管改革的持续深化为创新药审评提供了高效的制度保障。其次,中国医药产业研发投入持续增长,创新生态系统不断完善。第三,CDE通过药审云课堂、沟通交流会议等多种形式,为申请人提供了全方位的技术指导服务,2025年共举办药审云课堂12期、开展授课56讲、13万余人在线观看,有效提升了行业的研发水平和申报质量。
4.3 新兴治疗领域布局加速
CDE在2025年展现了在新兴治疗领域的前瞻性布局,特别是在细胞与基因治疗、创新疫苗、放射性药物等前沿领域取得了重要进展。截至2025年底,已有8款CAR-T细胞治疗药品在中国上市,用于治疗复发难治性淋巴瘤、白血病、多发性骨髓瘤等疾病。中国首款干细胞治疗药品获批用于移植物抗宿主病治疗,中国首款基因治疗药物获批用于血友病治疗,均为相关领域患者提供了新的治疗选择。
在创新疫苗领域,CDE通过牵头开展"人用疫苗新型佐剂的技术评价研究"课题,发布了《疫苗佐剂非临床研究技术指导原则》,首次制定了佐剂药学技术指南,完善了佐剂疫苗技术评价体系。2025年,多款含新型佐剂的IND疫苗品种获批,涵盖流感疫苗、呼吸道合胞病毒疫苗、带状疱疹疫苗、狂犬疫苗、结核疫苗等。在多糖结合疫苗领域,CDE制定了全球首个多糖结合疫苗药学开发全过程的通用型指导原则,有效填补了国内外空白,推动20个多糖结合疫苗进入临床。
放射性药物是CDE在2025年重点发力的另一新兴领域。全年批准放射性药品6个品种,其中3个品种通过优先审评审批程序加快上市。CDE制定了《放射性治疗药物申报上市临床风险管理计划技术指导原则》,为放射性药物临床研发提供了指导。随着核医学的快速发展,放射性药物在肿瘤诊断和治疗中的应用前景广阔,CDE的前瞻性布局将为中国在这一领域的发展奠定坚实基础。
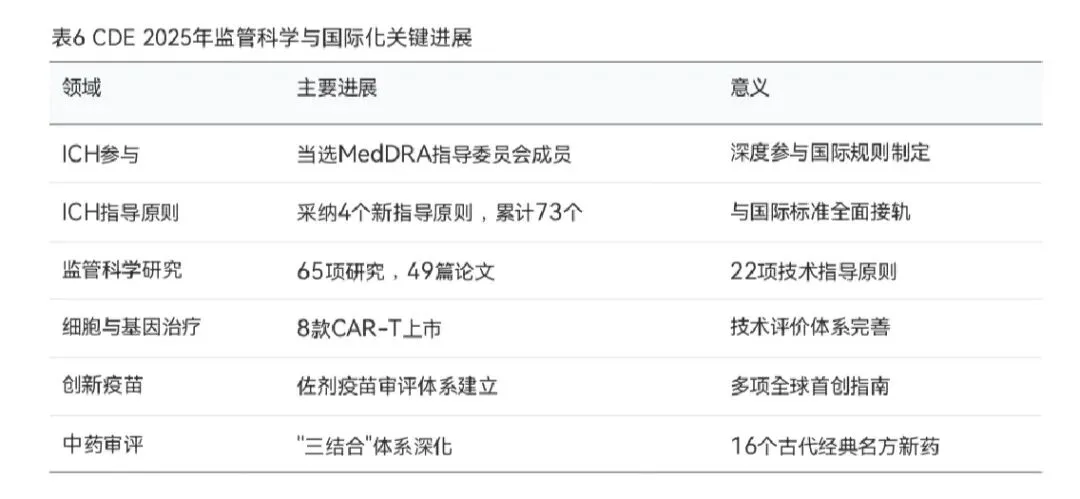
4.4 监管科学与国际化进程
CDE在2025年持续推进监管科学研究和国际化进程,取得了多项重要突破。在国际协调方面,国家药监局在2025年5月的ICH马德里会议上成功当选为ICH MedDRA指导委员会成员,这是指导委员会自2025年改组以来首次扩增成员,标志着中国药品监管将更加深入地参与国际规则制定。全年共选派83人参与48个ICH议题工作组的国际协调,参加643场电话会议,持续深入参与ICH指导原则的制修订。
在指导原则转化实施方面,2025年国家药监局采纳了4个新制定或修订的ICH指导原则,包括M14(真实世界数据安全性评估)、M11(临床电子结构化协调方案)、E6(R3)(药物临床试验质量管理规范)和E2D(R1)(上市后安全数据管理)。截至2025年底,国家药监局已累计采纳实施73个ICH指导原则,中国药品注册技术要求与国际标准的协调一致性不断提高。
在监管科学研究方面,CDE依托国家重点研发计划、国家药监局"中国药品监管科学行动计划"等平台,组织实施了65项药品监管科学研究,与117家产学研监等相关方持续互动交流。研究过程中发表了49篇学术论文,固化了22项技术指导原则和21项调研报告,形成了新的监管理念、工具和方法。特别是在新方法学(NAMs)领域,CDE与中检院联合涵盖产、学、研领域的7家机构共同推进类器官/器官芯片方法学的验证和适用场景探索,为替代传统动物实验、提高药物研发效率提供了科学依据。
五、结论与期待
5.1 主要结论
第一,中国药品审评效率已跃居全球前列。2025年NMPA批准的76个1类创新药,在数量上连续第二年超过FDA和EMA,年均复合增长率高达30.6%,充分反映了CDE审评审批制度改革的显著成效。多种加快上市程序的建立和完善,使得创新药从研发到上市的周期大幅缩短,有效提升了中国患者的用药可及性。
第二,三大监管机构各具特色和优势。NMPA的优势在于审评数量增长迅速、加快上市程序体系完善、中药审评独具特色;FDA的优势在于first-in-class药物占比高、罕见病用药审评经验丰富、审评标准全球引领;EMA的优势在于生物类似药审评全球领先、ATMP审评体系成熟、获益-风险评估审慎科学。三者形成了各具特色的监管模式。
第三,中国创新医药发展进入快车道。国产创新药占比持续提升,新机制新靶点药物不断涌现,细胞与基因治疗、创新疫苗、放射性药物等新兴领域布局加速,监管科学研究和国际化进程深入推进。中国正在从仿制药大国向创新药强国加速转型。
第四,审评质量与创新激励的平衡仍是共同挑战。三大监管机构都在努力加快审评速度的同时,确保审评质量和药品安全。FDA 2025年批准数量的下降、EMA发布的7项消极意见,都反映了监管机构在加速审评与严格标准之间寻求平衡的审慎态度。CDE在快速增加批准数量的同时,也需要持续加强审评质量管理体系建设。
5.2 期待
一是持续优化审评程序体系。
在现有突破性治疗药物程序、附条件批准、优先审评审批的基础上,进一步探索"滚动审评"、"实时审评"等创新机制,提高审评效率。同时,完善30日临床试验审评通道的实施效果评估,适时扩大适用范围,为创新药临床开发提供更大的便利。
二是加强源头创新支持力度。
虽然中国创新药批准数量已位居全球前列,但first-in-class药物占比(2025年约14.5%)仍低于FDA(约43%)。建议通过完善专利链接制度、数据保护制度等激励机制,加大对源头创新的支持力度,推动中国创新药从"跟跑"向"并跑"乃至"领跑"转变。
三是深化国际合作与互认。
以ICH指导原则转化实施为抓手,进一步深化与FDA、EMA等国际监管机构的合作,探索药品审评的国际互认机制。特别是在生物类似药、ATMP等领域,可以参考EMA的成熟经验,完善中国的技术评价体系。同时,积极推动中国监管科学研究成果向国际标准转化,提升中国在国际药品监管领域的话语权。
四是强化审评质量与风险管理。
在审评数量快速增长的同时,必须确保审评质量的同步提升。建议进一步完善审评质量管理体系,加强审评人员的培训和能力建设,建立更加科学的审评标准体系。同时,加强药品上市后监管,完善药物警戒体系,确保公众用药安全。
五是促进区域审评均衡发展。
CDE已在长三角、大湾区、京津冀、西南等地设立审评检查分中心,建议进一步发挥分中心的区域服务功能,加强与地方药监部门的协同联动,促进区域医药产业的均衡发展。同时,持续开展面向区域的药品注册技术培训,提升地方企业的研发水平和申报质量。
参考文献
1. 国家药品监督管理局药品审评中心. 2025年度药品审评报告[R]. 北京: 国家药监局, 2026.
2. U.S. Food and Drug Administration. Novel Drug Approvals for 2025[EB/OL]. FDA CDER, 2026.
3. European Medicines Agency. Human Medicines Highlights 2025[R]. Amsterdam: EMA, 2026.
4. Mullard A. 2025 FDA approvals[J]. Nature Reviews Drug Discovery, 2026, 25: 81-87.
5. Albericio F, et al. The Pharmaceutical Industry in 2025: An Analysis of FDA Drug Approvals from the Perspective of Molecules[J]. PMC, 2025.
6. FDCELL. EMA 2025 Human Medicines Approvals: Key Regulatory Highlights[EB/OL]. 2026.
7. Kayki-Mutlu G, et al. A year in pharmacology: new drugs approved by the US Food and Drug Administration in 2024[J]. Naunyn-Schmiedeberg's Archives of Pharmacology, 2025.
8. Papapetropoulos A, et al. Novel drugs approved by the EMA, the FDA, and the MHRA in 2023: a year in review[J]. British Journal of Pharmacology, 2024, 181: 1553-1575.
9. Zhou Y, et al. Global first-in-class drugs approved in 2023-2024: Breakthroughs and insights[J]. The Innovation, 2025.
10. JAMA Health Forum. Expedited Approval of Urgently Needed Drugs in China[J]. JAMA, 2025.