


原文翻译
所有报告基因的设计原则为:确保表达的 mRNA(包括启动子区域与终止信号)不含任何 DRACH 基序或 mcm⁵s²U 依赖性密码子,除非是实验中刻意引入的序列。所有元件均由南京金斯瑞(GenScript)合成,并通过标准分子克隆或吉布森组装(Gibson assembly)方法克隆至 pcDNA3.1 (+) 载体(赛默飞世尔)中。
研究使用mCherry 基因作为表达内参:该基因已通过同义密码子替换,移除了所有含 m⁶A 的 DRACH 基序与 mcm⁵s²U 依赖性密码子。
在 CMV 启动子控制下,克隆N 端带 V5 标签、不含 DRACH 基序与 mcm⁵s²U 依赖性密码子的 mNeonGreen 序列,其后连接一段由12 个重复 DRACH 基序或12 个重复 DRAGH 基序组成的片段。针对每个密码子特异性片段,选取GLORI 数据(Liu 等,文献 13)中显示 m⁶A 修饰水平最高、且包含目标密码子的 3 种 DRACH 序列基序,将其串联后再重复 4 次,最终构成 12× 重复单元。
为构建3′UTR 报告基因,将 mNeonGreen 与 12×DRACH 基序之间的CGA 密码子替换为TGA 终止密码子。
该方法详细介绍(原理 + 设计 + 用途)
1. 方法核心目的
严格控制变量,精准验证:mRNA 上不同位置的 m⁶A,是否会导致降解差异。只改变 m⁶A 位置 / 有无,不改变其他任何可能影响翻译、降解的序列。
2. 报告基因设计的 4 个关键原则(原文严格遵守)
(1)完全剔除背景干扰序列
载体、启动子、终止子、荧光蛋白全部不含 DRACH(避免自发 m⁶A 修饰) 全部不含mcm⁵s²U 依赖性密码子(避免 tRNA 修饰干扰) 目的:只有实验插入的 DRACH 能被 m⁶A 修饰,无背景噪音
(2)内参对照:mCherry
同样做了同义密码子全替换,无 DRACH、无 mcm⁵s²U 依赖密码子 用于校正转染效率、RNA 提取效率、荧光表达差异 保证降解速率的变化是目标报告基因的变化,不是系统误差
(3)核心报告单元:mNeonGreen + 12×DRACH/DRAGH
mNeonGreen:无 DRACH、无 mcm⁵s²U 依赖密码子 12×DRACH:可被 METTL3 甲基化,产生 m⁶A 12×DRAGH:DRACH 突变体,不能被甲基化,作为负对照 12 个重复:强化 m⁶A 信号,让降解差异更易被检测
(4)单碱基突变实现位置切换
- CDS 报告基因:mNeonGreen 后是
- 3′UTR 报告基因:仅把 CGA →
两个报告基因几乎完全一样,只差 1 个碱基→ 完美控制变量:唯一变量是 m⁶A 在 CDS 还是 3′UTR
3. 密码子特异性设计(GLORI 数据指导)
依据 GLORI 单碱基绝对定量 m⁶A 数据 挑选m⁶A 修饰率最高的 3 种 DRACH 串联并重复 4 次 → 12× 目的:让报告基因的 m⁶A 修饰强、稳定、可重复
4. 最终得到 3 组关键报告基因
- CDS-m⁶A:DRACH 在编码区 → 可甲基化
- 3′UTR-m⁶A:DRACH 在 3′非翻译区 → 可甲基化
- CDS-no m⁶A:DRAGH 在编码区 → 不可甲基化(负对照)
5. 该方法在本文中的科学作用
直接因果证明:CDS 中的 m⁶A 才会加速 mRNA 降解 排除序列长度、密码子偏好、RNA 结构等所有混淆因素 是本文从组学相关性 → 实验因果的关键支撑实验
结果展示:
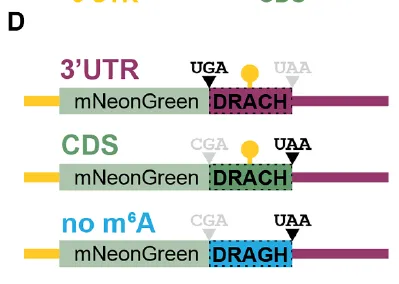
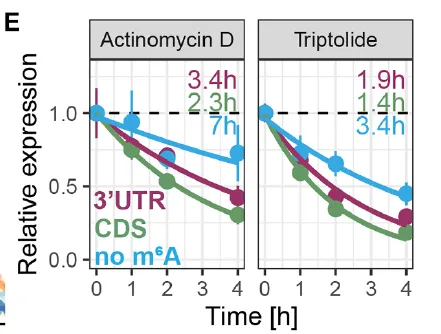
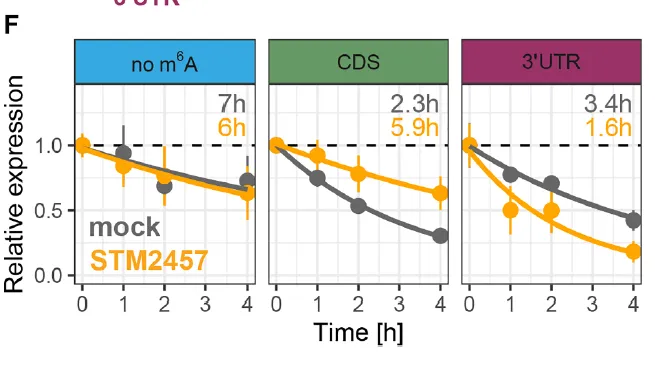
(D) 用于检测 m⁶A 位置效应的报告基因载体。m⁶A 修饰的 DRACH 片段分别位于 3′UTR(紫色)、CDS(绿色),或 CDS 中突变为不可甲基化的 DRAGH 片段(蓝色)。(E) 经放线菌素 D(左)或雷公藤内酯(右)关闭转录后,HEK293T 细胞中报告基因 mRNA 的降解。颜色方案同 (D)。(F) 经模拟处理(灰色)或 METTL3 抑制剂 STM2457 处理(橙色)的 HEK293T 细胞中报告基因 mRNA 的降解。