



关注康薪KANGLIFE,获取更多IVD及临床法规最新资讯

康薪咨询
KANGLIFE CONSULTING

近日,美国食品药品监督管理局(FDA)于2026年4月2日更新了2025年第一季度510(k)第三方审查报告,对比呈现了2023年至2024年MDUFA(Medical Device User Fee Amendments,医疗器械用户付费修正案)的执行情况。本期将对此报告进行详细解读,并洞悉FDA 510(k)审核的最新进展及动向。
01
MDUFA框架概述
MDUFA(Medical Device User Fee Amendments,医疗器械用户付费修正案)是美国国会授权美国食品药品监督管理局(FDA)向医疗器械企业收取审评费用的制度安排。作为对等机制,FDA需在约定时限内完成审评,并持续提升审评效率与透明度。
在MDUFA V周期内,FDA通过定期发布绩效报告(Performance Reports),对外披露包括510(k)审查在内的关键指标。同时,报告亦涵盖第三方审查项目(Third Party Review Program)的相关数据,如审查周期、补充信息请求情况等,从而提升行业对不同审查路径的可预期性。
该机制本质上形成了“第三方技术评估 + FDA最终裁定”的双层审查模式。
02
510(k)第三方审查计划背景
510(k)第三方审查计划,正式名称为 “认证机构计划”(Accredited Persons Program),由1997年《FDA现代化法案》(FDAMA,FDA Modernization Act)设立,旨在提升低至中等风险医疗器械的审查效率。
其核心机制包括:
FDA认可具备资质的第三方审核机构(3PROs, Third Party Review Organizations)
授权其对符合条件的510(k)申请进行技术审查
第三方机构提出实质等效性判断建议(SE实质等效/NSE非实质等效)
FDA基于审查结果作出最终监管决定
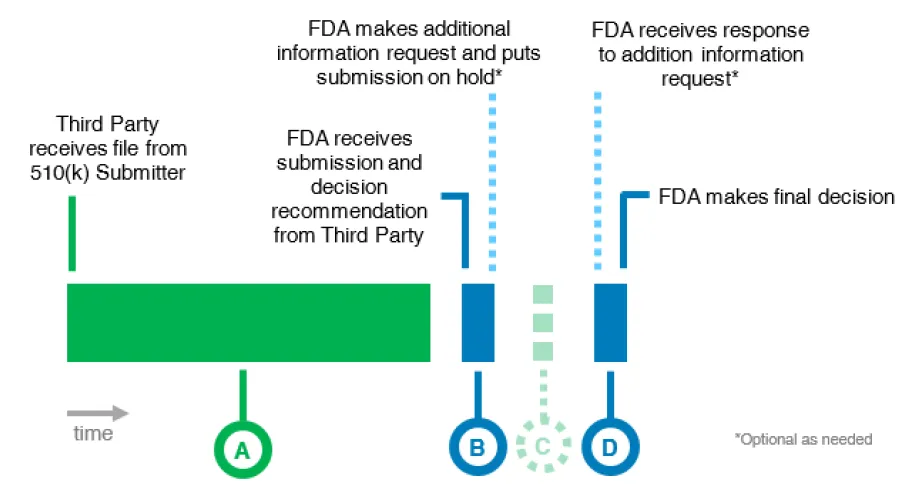
审查流程:
阶段A: 第三方审查机构从510(k)提交者处接收文件,对文件进行审查,并将文件及其决定建议发送给FDA。
阶段B:FDA审查提交材料,确保第三方审查机构已提供做出最终决定所需的全部信息。如需更多信息,FDA提出补充信息要求,通知第三方审查机构,并将该提交申请暂停 (Hold) 处理。
阶段C:若FDA提出补充信息要求,第三方审查机构审查FDA的要求并通知510(k)提交者。第三方审查机构对FDA的不足之处作出回应,根据需要更新审查备忘录和提交材料。在FDA收到对补充信息要求的完整回复之前,该提交申请视为暂停 (Hold) 状态。
阶段D: FDA审查补充信息并做出最终决定。
该机制本质上形成了“第三方技术评估 + FDA最终裁定”的双层审查模式。
(*图片来源:FDA, 《FDA 510(k) Third Party Performance Report FY26, Q1》)
03
FY2023–FY2026关键数据趋势
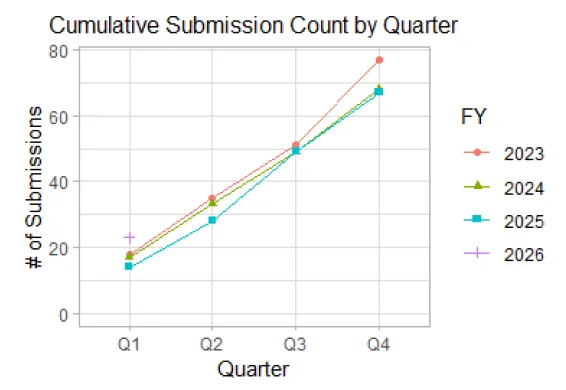
2023-2026年各财年按季度统计的第三方510(k)累计受理数量汇总:
(*图片来源:FDA, 《FDA 510(k) Third Party Performance Report FY26, Q1》)
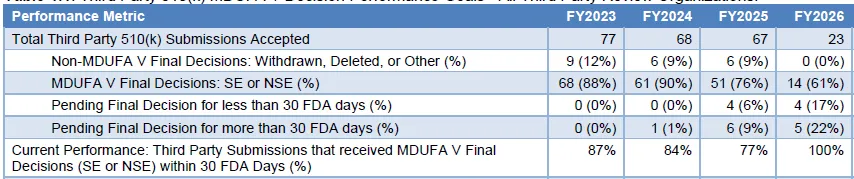
所有第三方审查机构的MDUFA V第三方510(k)决策绩效目标示意:
(*图片来源:FDA, 《FDA 510(k) Third Party Performance Report FY26, Q1》; 注:以上数据截至2026年Q1)
01 | 申请规模变化
数据显示,获得最终决定的第三方510(k)申请数量从FY2023出现整体收缩趋势。
这一变化可能受到多重因素影响,包括但不限于:
第三方审查适用产品范围的限制;
企业申报路径选择策略变化;
不同审查路径之间的效率与确定性权衡。
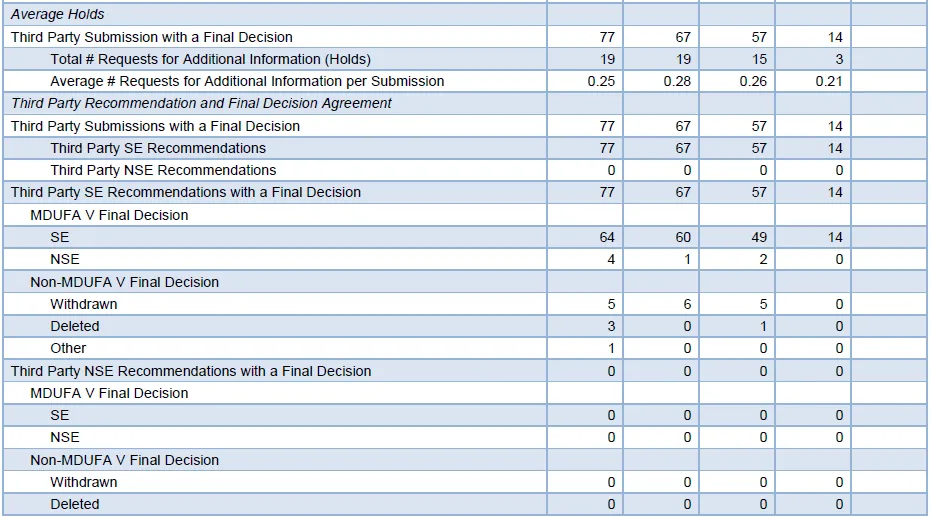
02 | 补充信息请求(Additional Information)与审查互动
同期数据显示:
补充信息请求总次数显著下降
平均每案发补次数降至较低水平
审查暂停(hold)时间明显缩短
该趋势可能反映:
标准化申报工具(如eSTAR)的应用提升了申报质量
第三方审查机构在正式提交前的预审查与沟通作用增强
进入第三方路径的案件整体复杂度相对可控
总体来看,审查互动频次的下降更可能体现流程优化,而非单一因素驱动。
(*图片来源:FDA, 《FDA 510(k) Third Party Performance Report FY26, Q1》; 注:以上数据截至2026年Q1)
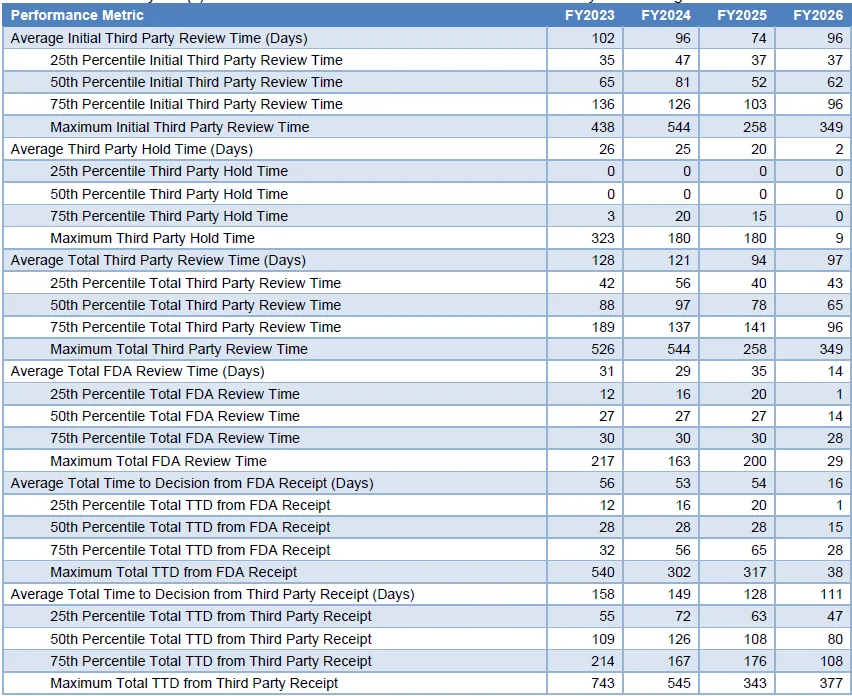
03 | 审查时效变化
从时间维度看:
FY2026第三方平均初审时间为96天,较FY2025增加22天
第三方审查阶段平均暂停时间为2天
第三方审查平均总时间为97天,较FY2025略有增加
FDA平均审查时间为14天
FDA收件至决定时间(Total Time to Decision from FDA Receipt)为16天
第三方收件至决定时间(Total Time to Decision from Third Party Receipt)为111天
(*图片来源:FDA, 《FDA 510(k) Third Party Performance Report FY26, Q1》; 注:以上数据截至2026年Q1)
综合来看,FY2026第三方审查流程呈现出“审查重心前移至第三方阶段、FDA审查与决策时间压缩”的特征。
在第三方初审时间延长的同时,补充信息请求及暂停时间大幅减少,使整体流程更加线性化。尽管第三方阶段耗时略有增加,但由于审查往返减少,整体收件至决定时间整体所见,反映出系统性流程效率的提升。
04 | 审查结果结构
根据2026年Q1数据记录,我们推测:
第三方审查主要适用于低至中等风险、路径明确的产品;
较复杂或存在不确定性的申请更可能直接进入FDA常规审查流程。
*注意:由于报告仅更新至2026年Q1,并非代表2026年整体情况,我们仍将持续关注2026年后三季度的持续更新。
04
MDUFA监管与行业意义
01 | 透明度持续提升
在MDUFA框架下,FDA通过持续披露绩效数据,提高了第三方审查项目的透明度,使企业能够基于实际数据评估不同审查路径的效率与适用性。
02 | 审查效率与适用范围的再平衡
尽管整体审查周期有所缩短,但申请量下降可能由以下因素共同驱动:
第三方路径逐步向特定类型产品集中;
企业在路径选择上更加审慎;
审查效率与适用范围之间形成新的平衡关系。
03 | 项目定位的演变趋势
从数据趋势看,第三方审查项目可能呈现以下演变方向:
更聚焦于低复杂度、标准明确的产品类型
强化“快速处理”能力而非扩大覆盖范围
在整体审评体系中承担补充性而非替代性角色
总结
FY2026数据显示,FDA 510(k)第三方审查项目呈现出“效率提升”与“规模收缩”并存的特征。在MDUFA V框架下,绩效数据披露机制有效提升了监管透明度。
与此同时,申请量下降及审查互动模式变化,反映出该项目在适用范围、案件结构及企业使用策略方面正在发生调整。整体来看,第三方审查路径仍是特定情境下的重要补充机制,但其角色正趋于更加聚焦与选择性使用
给医疗器械制造商和IVD制造商的建议:
第三方审查路径在特定条件下仍具备时间优势
但更适用于FDA明确规定的适用产品范围
在路径选择时建议重点评估,包括产品复杂度及风险等级、实质等效路径的明确性、审查时间与确定性需求
同时:
对于技术复杂度较高或存在不确定性的产品,仍建议优先考虑FDA直接审查路径
企业需持续关注第三方审查机构数量及活跃度变化对路径可行性的潜在影响
- 康薪咨询一站式美国市场准入解决方案 -
康薪咨询专注于全球市场准入及临床试验合规咨询服务,为大型、中小型及初创企业提供一站式、定制化的解决方案。
FDA产品分类及注册路径决策
FDA 510(k)预审核
FDA 510(k) 技术文件辅导
FDA QMSR 质量管理体搭建
上市后监管
美国当地代理等
我们致力于与医疗器械企业携手合作,助力其产品稳步走向全球市场。
*资料来源:FDA《FDA 510(k) Third Party Performance Report FY26, Q1》
*声明:本文章报告内容均为转载,仅用于分享,如涉及版权等问题,请随时与我们取得联系,我们将于第一时间进行修正,谢谢。
康薪咨询团队可承接欧洲多国、泰国、中国等多国医疗器械及IVD器械注册要求的临床实验研究项目(可出具伦理批件),可为IVD企业提供全生命周期的解决方案服务。
康薪KANGLIFE
业务范围
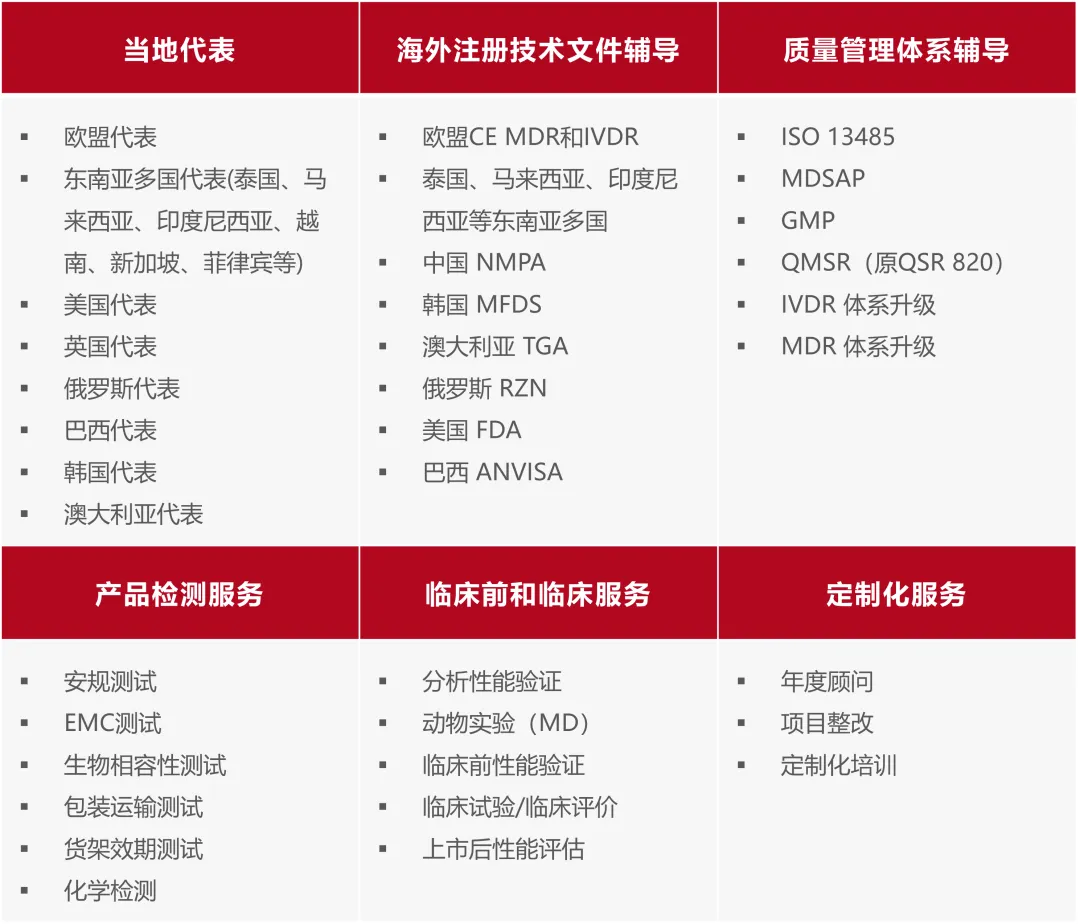
注册服务支持区域
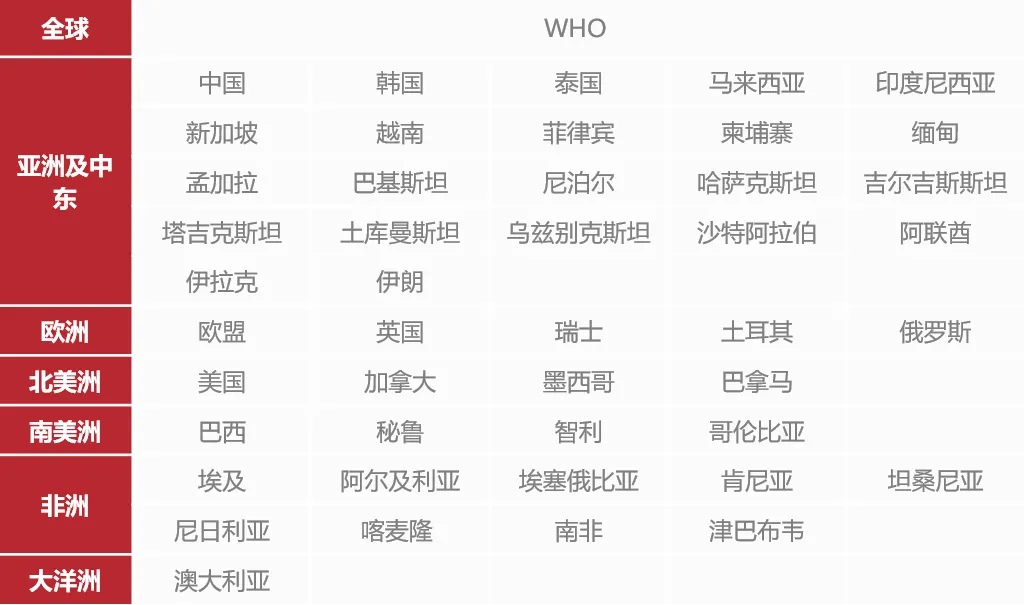
临床实验室资源

关于康薪咨询
ABOUT KANGLIFE
| 专业特色:
协同企业,共同定制出海战略服务。
| 核心文化:
协助国内外优秀体外诊断及医疗器械企业实现战略发展、培养合规人才,携手共筑中国企业出海愿景。
康薪咨询成立以来已与数百家企业强强联合,协同高效合作,提供定制化服务,成功获得多个欧美和东南亚市场准入资质。
推荐阅读
业务咨询
CONTACT US

华北区咨询顾问
Harold Hua

华东区咨询顾问
Joyce Hua

华南区咨询顾问
Fitter Cao


关注康薪咨询
Follow KANGLIFE
点击下方”阅读原文“,即刻与我们取得进一步联系!