



欢迎扫码进群交流

药品生产企业整改报告编订指南(征求意见稿)
一、目的
依据《药品注册管理办法》《药品生产监督管理办法》《药品检查管理办法(试行)》《药品医疗器械境外检查管理规定》等相关要求,对药品生产企业现场核查 / 检查(以下统称为 “检查”)缺陷整改报告的编订要求进行规范和统一,提高企业编订和监管部门审核效率,督促企业强化质量风险闭环管理,持续完善质量管理体系。
二、适用范围
本指南所指的整改报告是指药品生产企业接受药品监管部门组织的现场检查后,针对检查发现的缺陷,按照实际整改情况,编订的反映企业调查分析、风险评估、风险控制(含纠正措施和预防措施)、完成情况等内容以及整改证明材料的一系列文件和记录。
本指南适用于药品监管部门依据《药品注册管理办法》《药品生产监督管理办法》《药品检查管理办法(试行)》《药品医疗器械境外检查管理规定》等开展的以药品生产企业为检查对象、以药品生产质量管理规范(GMP)为检查标准的各类检查。
其他类型检查可参考使用。
三、基本要求
(一)整改责任
药品生产企业的法定代表人、企业负责人等高层管理人员,应当作好缺陷整改的组织策划,提供必要的人力资源和资金保障,协调缺陷相关部门密切沟通,明确职责和分工,积极落实整改要求,确保缺陷得到有效整改,产品和质量体系的风险得到有效控制,组织编写并按要求提交整改报告。
若为受托生产,药品上市许可持有人(以下简称 “持有人”)应当按照法规要求、持有人与受托生产企业质量协议和工作程序,履行相应整改责任。
(二)质量管理
检查缺陷的整改工作(含缺陷调查、风险评估、整改过程及审核、整改效果评估)应当纳入质量管理体系(如质量风险管理、偏差处理、变更控制、纠正措施和预防措施等),形成闭环管理。
(三)整改要求
缺陷调查和风险评估的深度、广度、范围和形式要与缺陷的风险水平相适应,风险控制、纠正措施和预防措施要及时有效,整改审核要严格谨慎,整改效果评价要客观充分。应当基于缺陷的风险水平进行全面或专项排查,举一反三,排查类似问题,及时进行整改,持续改进质量管理体系。(例如通过自检等方式开展)
(四)整改时限
检查缺陷应当按照各类检查程序要求在规定时限内完成整改。无法按期完成整改的,必须有充分的理由,应当制定切实可行、时限合理的整改计划,同时针对缺陷存在的风险,采取必要的风险控制措施。后续整改措施应按照计划执行。企业按照整改计划完成整改后,应当及时将整改情况形成补充整改报告报送派出检查单位,如有特殊情况需变更整改计划的,企业应当充分评估计划变更带来的影响,并经质量负责人审核批准后,报告派出检查单位。
派出检查单位在审核整改报告的过程中,认为需要企业进行补充整改的,补充整改时限以派出检查单位要求为准。
(五)编订原则
整改报告由正文和附件组成。
正文包括两部分。第一部分为整改概述,包括检查基本情况、整改基本情况;第二部分为整改详述,包含具体的缺陷描述、缺陷调查分析、风险评估、风险控制(含纠正措施和预防措施)、整改完成情况(含整改审核、效果评价)等内容,针对缺陷成因及风险评估情况,逐项描述风险控制措施及实施结果。
附件是正文内容的证明性材料。
整改报告内容应当真实完整、文字应当表述准确,逻辑清楚通顺,充分、如实反映企业的整改情况。
四、编写要求
(一)正文第一部分:整改概述
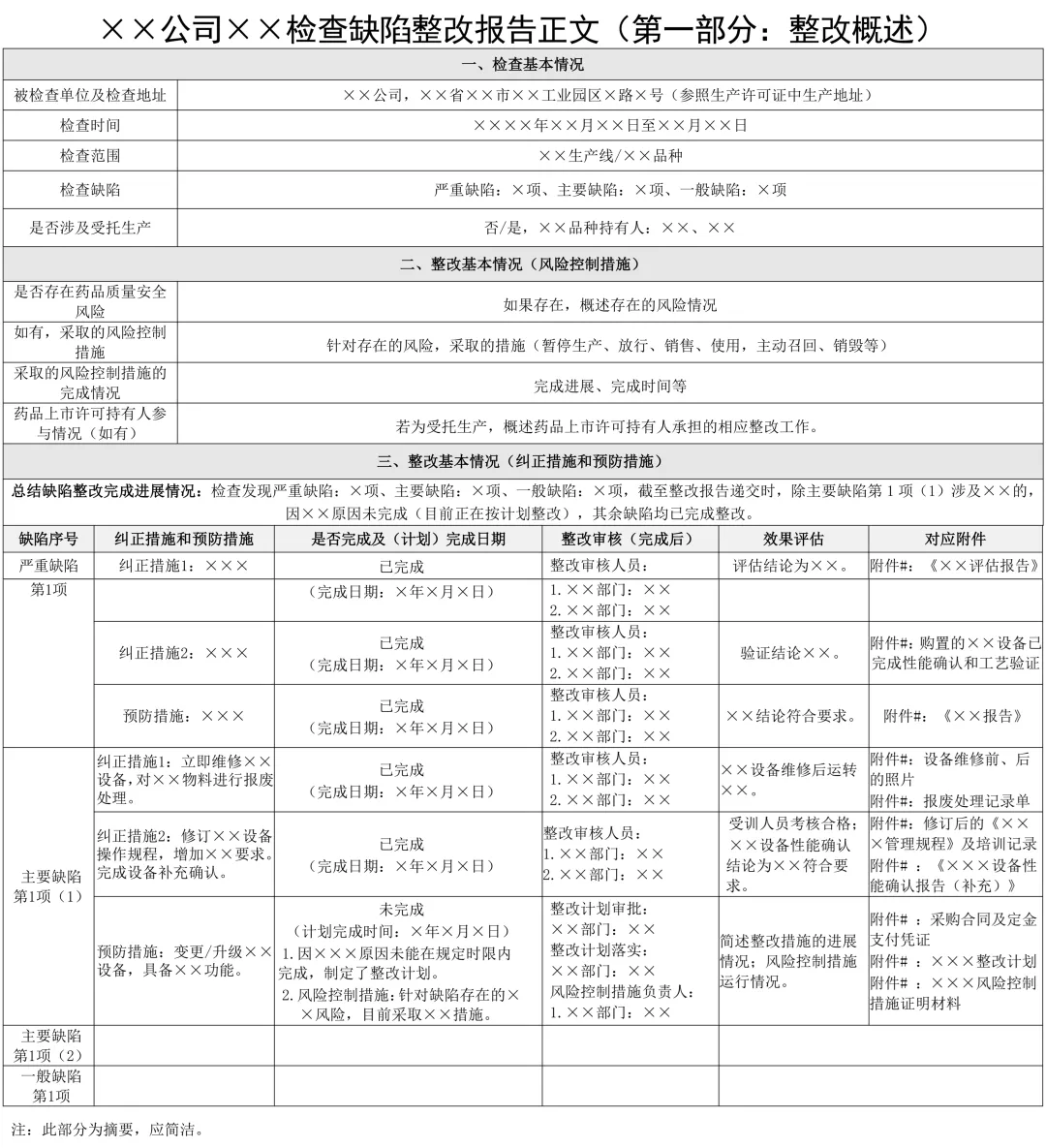
1.检查基本情况
包括被检查单位及地址、检查时间、检查范围、检查缺陷项目情况。
2.整改基本情况
(1)概述风险控制措施情况。若为受托生产,需概述持有人承担的相应整改工作(如有)。
(2)概述纠正措施和预防措施概述已完成整改情况,包括完成时间、整改效果评价。概述未完成整改情况,包括计划完成时间、跟进落实的人员。针对该缺陷存在的风险,整改完成前,是否制定了风险控制措施。
(二)正文第二部分:整改详述
1.缺陷描述
首先描述缺陷原文,其次对缺陷原文补充说明(如缺陷发生的时间、地点或部门、具体情节及相关人员等,若涉及产品 / 物料的,需明确产品 / 物料的批号、规格、数量等)。若缺陷原文描述已足够详细,则无需补充。若缺陷项含有多个独立小项,需按小项逐项描述,避免漏项。(下同)
2.调查分析
概述调查过程及结果,阐明缺陷发生的根本原因。调查的深度和形式应当与缺陷的风险水平相适应。企业需遵循科学、客观的原则,采用适宜的调查分析方法,明确缺陷发生的根本原因(无法确定根本原因时,应分析潜在原因)。
3.风险评估
概述风险评估的过程及方法、缺陷的风险水平以及其对药品质量或质量管理体系的影响。存在药品质量安全风险的,应当详述。
结合调查分析的结果,基于 ICH Q9 质量管理的原则,可采用适宜的风险管理方法与工具,综合评估缺陷对药品质量安全生产的不良影响,以及其对质量管理体系单个环节、相关环节乃至整个质量管理体系的影响。质量风险管理实施过程的深度、正式程度和文件化程度都应当与风险水平相适应。必要时,应当结合工艺性能和质量监测趋势或其他来源的质量数据,开展产品质量风险评估。
4.风险控制
风险控制应当包括风险控制措施及纠正措施和预防措施。
(1)风险控制措施
企业整改期间应当主动结合调查过程中发现的药品质量安全风险,采取与缺陷的风险水平相匹配的风险控制措施,包括暂停生产、放行、销售、使用及主动召回、销毁等。
根据风险评估结果,对于存在药品质量安全风险的,除在此处描述已采取的风险控制措施,还应当同时明确责任部门和责任人,以及整改完成时间或合理的计划完成日期。
(2)纠正措施和预防措施
根据缺陷原因调查情况及风险评估结果,此处逐项描述制定的纠正措施和预防措施。
①纠正措施:为消除已发现的缺陷或其他非期望状况的原因所采取的措施,其目的是消除缺陷产生的原因,防止缺陷再次发生。
②预防措施:为消除潜在的缺陷或其他潜在的非期望状况的原因所采取的措施,是防止缺陷发生的措施,即为消除潜在的缺陷原因所采取的措施。
5.整改完成情况
整改完成情况应当包括整改审核、效果评价等内容。
逐项描述纠正措施和预防措施是否完成,效果评价(涉及稳定性考察 / 验证 / 确认 / 校准 / 风险评估工作的应当概述最终结论,以培训为主的整改措施应当概述培训效果,以修 / 制定文件落实整改措施的需概述修 / 制订的内容等)、完成日期和整改审核人(完成后)。
无法在规定时限内完成的整改措施,应当详述原因,提交切实可行、时限合理的整改计划,同时结合缺陷存在风险制定必要的风险控制措施,明确整改计划的审核人、批准人,跟进落实的责任部门、责任人和计划完成时间。
整改完成情况应当与提交的附件证明材料相对应。
(三)附件
提供正文内容的相关证明性资料,如风险评估报告、修订的文件或记录、培训材料、验证 / 确认方案和报告、校准证书、许可或备案文件、图纸、整改计划(仅未完成的措施需提供)等复印件,设备或现场照片、视频 / 录像等。
注意:同一个证明材料支持多个缺陷整改的,可仅提供一次,报告正文需明确说明具体索引情况。
五、格式要求
(一)纸质资料
1.内容
应当包括上述整改报告正文、附件证明材料(附件若为视频 / 录像,则仅需提供电子资料)。
2.语言
境外药品生产企业检查缺陷整改报告正文及附件应当包括中文版本和英文版本(或以中英文对照形式)。境内药品生产企业整改内容涉及外文文献或资料,需同时提供中文和英文版本。企业对翻译的准确性负责。
3.文字格式
(1)字体:中文宋体,英文 Times New Roman。
(2)字号:不小于小四号,表格内文字不小于小五号。
(3)字体颜色:黑色。
(4)行间距及页边距离
行间距:至少为单倍行距。
页边距离:在准备文本和表格的过程中应留出一定的页边距,以便文件能够用 A4 纸(图纸除外)印刷。左侧的页边距应足够宽,以便装订时不会遮挡住文中的内容。纵向页面:推荐左边距离不小于 3 厘米、上边距离不小于 2 厘米、其他边距不小于 1 厘米;横向页面:推荐上边距离不小于 3 厘米、右边距离不小于 2 厘米、其他边距不小于 1 厘米。
4.印章要求
整改报告正文、附件应当加盖药品生产企业公章(首页及骑缝章);境外药品生产企业整改报告及附件应当加盖境内责任人公章(首页及骑缝章)。加盖的公章(含电子印章)应当符合国家有关用章规定,并具有法律效力。
5.整理
整改报告正文和附件需分别制作目录并添加页码(从封面开始,有书写内容的页面均需添加)。
整改附件若不便添加页码,可仅制作目录,但须按缺陷顺序标记编号、命名,以便从正文快速索引,不同附件之间须以简易、明显的方式进行适当的标记或物理间隔,以便于快速区分和检索。
6.装订
整改报告正文及附件所用幅面一般采用国际标准 A4 型(图纸除外),单面或双面打印(为节约用纸,尽可能双面打印)。
(1)整改报告正文单独成册(一般不与附件合并),采用文件夹、长尾夹、胶装等方式装订(注意不得使用金属钉)。
(2)整改附件按照缺陷级别按顺序合并成册(如主要缺陷合并为册、一般缺陷合并为册),每册附件的厚度一般不大于 300 张,可采用打孔线、胶装、文件夹、档案夹等方式装订(注意不得使用金属钉),每册应加封面,注明:整改材料内容(如:主要缺陷第 1 项至主要缺陷第 2 项整改附件证明材料)、企业名称、检查品种 / 范围、检查时间、企业联系人信息、第 × 册 / 共 × 册。
(二)电子资料
1.制作
将上述纸质整改资料扫描制作成 pdf 文档。具备电子印章的企业,可以使用由源文件(如 WPS Word 文件)转化形成的 pdf 文档,并使用电子印章。
2.整理及命名
为满足检索、查询需要,整改报告需在关键信息的起始位置制作电子书签 / 标签,数量不得少于目录条数。
整改报告正文两部分需单独制作 pdf 文档。附件可合并成一个 pdf 文档也可单独提交。一个 pdf 文档对应一个附件时,pdf 文档命名至少包含附件编号和文件名称;多个附件合并为一个 pdf 文档时,需制作电子书签 / 标签,pdf 文档命名至少包含对应缺陷项级别序号及附件编号。
附件 pdf 文档放在不同文件夹时,文件夹命名需包含缺陷项序号。
3.电子传输介质
电子整改材料可通过 USB 闪存驱动器(即 U 盘)、光盘(一次性写入型,CD-R、DVD-R、DVD+R)或者其他网络形式(如电子邮箱、申请人之窗、网盘)提交。若使用光盘,每张光盘表面(非数据读取面)使用光盘标签笔清晰标注 “企业名称、检查时间、第 × 张 / 共 × 张” 等信息,光盘表面不得粘贴标签。光盘应当装入光盘硬盒中,光盘盒封面需标记 “光盘内容、企业名称、检查品种 / 范围、检查时间、企业联系人信息、第 × 张 / 共 × 张”。若使用 U 盘,请参照光盘做好标记和区分。
(三)其他注意事项
1.整改材料中提供的复印件应当与原件完全一致并保持完整、清晰。
2.为提高审核效率,整改涉及文件 / 记录修订的,提供的附件需以彩色水笔 / 高亮颜色将修订前、后的内容进行标记。
3.电子文档的拆分及合并需控制在传输介质(电子邮箱、申请人之窗、网盘等)允许范围(如单个 pdf 文档不得超过 100M 或 50M,具体以派出检查单位要求为准)之内。
六、报送要求
缺陷整改材料应当以药品生产企业正式公文(包含发文字号等)提交,随附整改报告。若为受托生产,整改报告需由受托生产企业及相关品种持有人共同确认后提交。
上述整改资料份数、递交渠道和递交形式(纸质 / 电子)应当以派出检查单位的要求为准。一般情况,对于严重及主要缺陷,企业应当提供完整的附件证明资料;对于一般缺陷,若正文描述详细且已足够反映实际整改情况,可无需提交附件证明资料。
七、补充整改报告
派出检查单位在审核整改期间,提出补充整改要求的,补充整改报告的编订应当参考本指南,编订时应当重点围绕补充事项,可对整改报告体例进行适当调整或简化。派出检查单位对补充整改有其他意见的,以派出检查单位意见为准。
八、参考样式
整改报告正文参考样式见附件。
参考样式是整改报告的一种呈现形式,企业可在本指南的基础上采用其他等同或更优的形式,整改报告内容应当涵盖《药品检查管理办法(试行)》相关要求。
附件 1×× 公司 ×× 检查缺陷整改报告正文(第一部分:整改概述)
附件 2×× 公司 ×× 检查缺陷整改报告正文(第二部分:整改详述)

1.严重缺陷
同后续主要缺陷。若药品监管部门有其他要求,应按照监管部门要求执行。
2.主要缺陷
2.1 主要缺陷第 1 项
风险及变更管理存在以下问题:(GMP 正文 第 243 条)
2.1.1 主要缺陷第 1 项(1)
2.1.1.1 缺陷描述
缺陷原文:(1)针对 ××× 供应商变更风险评估缺少……。
补充说明:×××(另起一段,描述对缺陷原文的补充,如有)
2.1.1.2 调查分析
概述调查过程,同时明确缺陷发生的根本原因。
2.1.1.3 风险评估
概述风险评估的过程及方法、缺陷风险级别以及其对药品质量或质量管理体系的影响。存在药品质量安全风险的,应当详述。
2.1.1.4 风险控制
(1)风险控制措施
根据风险评估结果,对于存在药品质量安全风险的,此处描述已采取的风险控制措施,同时明确相关的责任部门和责任人,明确完成时间或合理的计划完成日期。
(2)纠正措施
(3)预防措施
2.1.1.5 整改完成情况
逐项描述纠正措施和预防措施是否完成,效果评价(涉及验证 / 确认 / 校准 / 风险评估工作的应当概述最终结论;以培训为整改措施应当概述培训效果,以修 / 制定文件落实整改措施的需概述修 / 制订的内容等)、完成日期和整改审核人(完成后)。
无法在规定时限内完成的整改措施,应当详述原因,提交切实可行的整改计划,同时结合缺陷存在风险制定必要的风险控制措施,明确整改计划的审核人、批准人,跟进落实的责任部门、责任人和计划完成时间。整改完成情况应当与提交的附件证明材料相对应。
示例:
情形一:
上述措施均均已完成,其中修 / 制定的文件对相关人员开展了培训。详见:
(1)附件 #:修订后的《××× 管理规程》及培训记录,修订的文件增加了 ××,受训人员考核合格。(完成日期:× 年 × 月 × 日,整改审核人员:××)
(2)附件 #:《××× 设备性能确认报告(补充)》,设备性能确认结论为 ×× 符合要求。(完成日期:× 年 × 月 × 日,整改审核人员:××)
情形二:
除 ×× 措施外,其他均已完成,其中修 / 制定的文件对相关人员开展了培训。详见:
已完成:
(1)附件 #:修订后的《××× 管理规程》及培训记录,修订的文件增加了 ××,受训人员考核合格。(完成日期:× 年 × 月 × 日,整改审核人员:××)
(2)附件 #:《××× 设备性能确认报告(补充)》,设备性能确认结论为 ×× 符合要求。(完成日期:× 年 × 月 × 日,整改审核人员:××)
未完成:
(1)变更 ××× 设备。因 ××× 原因未能在规定时限内完成,制定了整改计划,具体包括:将于 × 年 × 月 × 日之前完成 ××× 工作,× 年 × 月 × 日之前完成 ××× 工作……,详见附件 #:《××× 整改计划》。整改计划审核人:×××,批准人:×××。计划完成日期:× 年 × 月 × 日,负责落实部门:×××,负责人:×××。
(2)风险控制措施:针对缺陷存在的 ×× 风险,目前采取的 ××× 措施,负责部门:×××,负责人:×××。证明材料见附件 #:《××× 风险控制措施》。
2.1.2 主要缺陷第 1 项(2)
2.1.2.1 缺陷描述
缺陷原文:(2)针对 ××× 供应商变更风险评估缺少……
补充说明:×××(另起一段,描述对缺陷原文的补充,如有)
2.1.2.2 调查分析
2.1.2.3 风险评估
2.1.2.4 风险控制
2.1.2.5 整改完成情况
2.2 主要缺陷第 2 项
3.一般缺陷
参考主要缺陷。若正文描述详细且已足够反映实际整改情况,可无需提交附件证明资料

大会名称| CPI·2026中国制药工业大会(杭州站)暨中国医药营销新资源论坛
大会时间 |2026年3月5-6日
大会地点 | 杭州和达希尔顿逸林酒店(浙江省杭州市钱塘区金沙大道600号)


大会名称| CPI·2026中国制药工业大会(杭州站)暨中国医药营销新资源论坛
签到时间 |2026年3月5日/6日8:00签到地点 | 杭州·和达希尔顿逸林酒店1楼签到处(请在完成签到后随身佩戴参会证,它是您参会的唯一有效证件,请妥善保管,遗失不补。)
大会地点 | 杭州和达希尔顿逸林酒店(浙江省杭州市钱塘区金沙大道600号)




药品监管政策动向高惠君 上海市食品药品安全研究会副会长、教授级高级工程师(正高级)
药品集采机制及医保政策的市场策略分析张廷杰 风云药谈创始人
项目推介
轻松一刻
新政下药企转型创新药研发政策解读王宗敏 江苏省药品监督管理局原药品注册管理处处长
药品上市后变更管理法规进展及要求吴浩上海药品审评核查中心,原学术委员会主任

药物杂质分析与控制杨永健 上海市食品药品检验研究院 原首席专家
药品微生物实验室质量管理及常见问题分析李辉 原国家药品监督管理局药品微生物检测技术重点实验室
轻松一刻
我国制药用水系统的最新法规动向与核查要求张功臣 医药行业专家、工信部领军人才
药品生产过程中的微生物控制王顺强 浙江天元生物药业有限公司生产副总经理

无菌制剂出口:体系优化与FDA飞检管理李华锋 正大天晴药业集团股份有限公司质量负责人、质量受权人
基于风险的无菌生产环境监控Barry Prabhu Novatek International, 亚太区总监
《药品受托生产监督管理》实施策略刘德富 联邦制药集团质量总监
轻松一刻
实施生命周期方法工艺验证的目的及实施策略牛萍 上海可立特技术有限公司创始人、总经理
药品生产现场核查流程及把控要点胡淼 原国家药品生产检查组长、制药产业学院特聘教授

如何正确理解和落实2025年版《中国药典》药包材标准体系俞辉 原浙江省食品药品检验研究院药品包装材料所所长
2025版《中国药典》四部通则2341方法解读和完整解决方案马海建 上海安谱实验科技股份有限公司 质量副总监
2025年版《中国药典》分析方法验证指导原则解读杨伟峰 原浙江省食品药品检验研究院化药所副所长,主任药师



透皮制剂创新趋势及开发策略赵江琼 赫力昂(中国)有限公司新品开发负责人
溶剂型贴剂生产工艺及关键技术钟元龙 楚天科技联席总裁、中央技术研究院常务院
外用制剂IVRT&IVPT研究策略及案例分享段云剑 深圳市华溶分析仪器有限公司技术总监
轻松一刻
透皮制剂IVPT的研究与实践郑枫 中国药科大学生物分析教授,现任CFDI核查专家
白发的药物靶点思考宋秀祖 教授,主任医师, 博士研究生导师,杭州市第三人民医院副院长,毛发医学研究中心主任

改良型新药:仿制药企业转型跳板郭新峰 南京循证生物科技有限公司创始人
药械协同创新:定制化给药器械在改良新药与复杂制剂中的应用与趋势孙文斌 杭州钱塘隆越生物科技有限公司开发技术部副总监
批文项目发布
轻松一刻
长效注射剂开发重难点剖析及案例分享岳占国 玻思韬纳米制剂总监
创新型吸入药物的研究与开发陈东浩 畅溪制药创始人

基于临床需求的改良型新药立项策略与实施案例江波 浙江大学医学院附属第二医院,机构办公室主任
3D打印在药物制剂领域的应用
邓飞黄 南京三迭纪医药科技有限公司 技术副总裁
轻松一刻
GLP-1多肽药物新剂型的开发策略探索郭晓迪 原浙江华海药业股份有限公司首席科学家
改良型新药的脱困与出海战略决策
王龙 SILRICH 注册和商务副总裁

透皮半固体制剂开发策略、质量研究及前沿计算科学应用综合研究王咏 浙江仙琚制药制剂研究院院长
透皮制剂质量研究应关注的问题杨永健 上海市食品药品检验研究院 原首席专家
透皮制剂的BE研究及临床指导郑恒 国家药品审评中心专家, 湖北省药监局核查专家,武汉市同济医院教授



中药注册政策与技术体系演进和趋势樊敏伟 国家中药制药工程技术研究中心副主任
医疗机构中药制剂新药转化策略与实践倪文澎 江苏省中医院制剂研发中心特聘首席专家
轻松一刻
基于拟人化模型的中药大健康产品研发陈素红 浙江中医药大学中医药科学院副院长、中药与健康产品研究院院长、二级研究员
中药复方制剂的传承与创新石森林 浙江中医药大学二级教授、浙八味研究所所长
数智赋能中医药诊疗技术研究洪燕龙 上海中医药大学 上海中医健康服务协同创新中心常务副主任、中药现代制剂技术教育部工程研究中心主任
中药注射剂“689”质量研究房强强 上海诗丹德标准技术服务有限公司,副经理
轻松一刻
同名同方药开发技术浅析李芳娜 谱尼生物医药中药研发中心负责人
中药复方申报现状及立项探讨胡江宁 浙江康恩贝制药股份有限公司药品研发中心执行总经理
经典名方的开发与市场前景
罗兴洪 转化医学与创新药物技术研究所 常务副所长

全产业链模式创新培养兽药行业新质生产力祝兴林 杭州和泽动保科技有限公司 总经理
宠物医药行业现状与研发趋势冯秀娟 金盾药业副总经理、科研团队首席专家
轻松一刻
宠物药物制剂多样化创新柯学 中国药科大学药学院副院长
创新性宠物原研医药与酶解功能产品开发与应用黄益波 立生生物创始人、董事长
人用药物的宠物适应症拓展策略与案例嘉宾行程确认中



做药20年,只有方向没有答案戴文杰 逍药圈主理人
模式革新——药企与代理商进入合规新时代冯鹤 国际注册会计师药企合规导师
茶歇与交流
集采与价格治理之下,药品营销逻辑解析唐锦 山东海山药业总经理
药械组合——用临床价值撬动百亿市场聂薇 博士,杭州畅溪制药
场景革命—— 换一种方式卖药胡江丽 医途科技联合创始人
实操案例解读药企合规模式与风险防范魏文龙 汇众聚诚董事长
三大案例揭示从核心打破医药行业内卷蔡华虎 满天星商学院产业研究院执行院长
药企视觉系统成构建企业品牌资产徐莎莎 药融圈视觉系统总监
“星光奖”年度包装设计大赛 颁奖仪式蔡华虎 满天星商学院产业研究院执行院长

开幕致辞
突围跨境:药企跨境电商的现状,策略与能力
陈志伟 广州健民医药连锁有限公司副总经理
平台赋能—— 京东国际的入驻与发展陈晓岚 京东健康跨境药品业务招商负责人
跨境电商+院外4.0:驱动药品第二增长曲线的营销革命柯小芳 药联众创始人
产品资源—— 跨境电商品类分析与基本货盘搭建吴华 华姐就药说主理人
金融支持—— 跨境电商供应链金融介绍叶路英 海南鑫富通
实操落地—— 医药电商的顺势而为与逆势增长孙亮亮 谁与医药董事长
跨境转型——一个律师转型做医药跨境电商的故事罗正宇 老挝康宁制药董事长
进口跨境医药与海南的机遇郭耀华 美极屋创始人 海南省电子商务协会跨境电商专委会主任
圆桌论坛——如何顺利完成跨境电商的布局?
1. 工业药企的渠道布局
2. 跨境业务的注意事项
3. 代理商的转型发展
4. 传统电商向跨境电商发展的路径


大会开放若干个展位:
更多现场展位火热抢订中,可为CRO/CDMO/仪器设备商/咨询服务等企业提供多形式的宣传展示:论坛承包、主题演讲、会场展位、项目路演、立屏展架、会刊彩页、挂牌、资料袋宣传、晚宴冠名、笔记本、茶歇赞助宣传等。
商务合作福利、收费标准等详情请咨询:
王女士 13588474548
周女士 15858667450
张女士 15005862516
周女士 13983785571

1. 制药企业董事长、总经理等决策层;
2. 制药企业研发、立项、临床总监、经理、研究员等人员;
3. 药学研发机构负责人;
4. 临床试验机构负责人;
5. MAH B证企业相关负责人;
6. 药物安全性评价机构、药物研发咨询机构相关人员;
7. 药企QA/QC/生产/注册等人员;
8. 医药科研院所、药检单位和医药高等院校中从事药物研究的相关人员;
9. 商务BD负责人;
10. 投资机构相关人员;
注册报名:大会限时免费(人数限制,先注册先得,本次报名为预登记报名,报名审核通过后为报名成功!),食宿自理。
参会报名咨询:
崔女士 13018913819 (微信同号)
文女士 18868801402 (微信同号)

扫码立即报名