


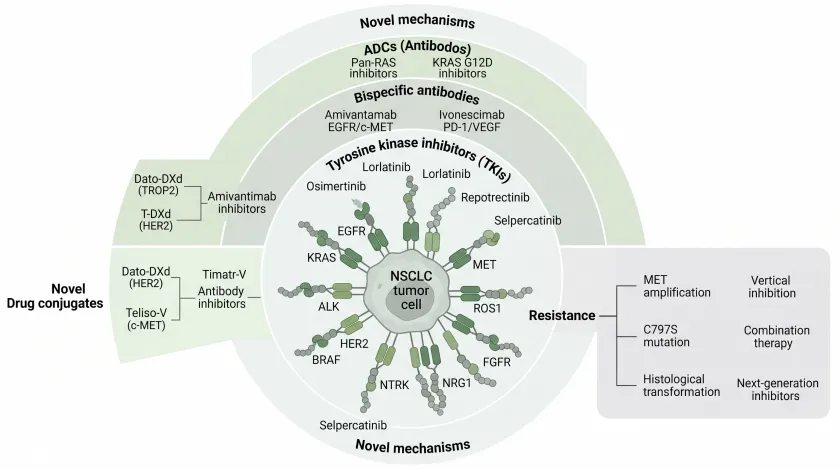
NSCLC靶向治疗全景机制示意图
图注:非小细胞肺癌(NSCLC)靶向治疗全景图。中央为NSCLC肿瘤细胞,外围展示主要可靶向驱动基因(EGFR、KRAS、MET、ALK、ROS1、RET、HER2、BRAF、NTRK、NRG1、FGFR)及其对应治疗药物分类:酪氨酸激酶抑制剂(TKI)、双特异性抗体、抗体药物偶联物(ADC)以及新型作用机制药物。右侧展示主要耐药机制(MET扩增、C797S突变、组织学转化)及克服策略(垂直抑制、联合治疗、新一代抑制剂)。
摘要
研究背景与目的
2024至2026年间,晚期非小细胞肺癌(non-small cell lung cancer, NSCLC)靶向治疗领域经历了前所未有的变革。美国食品药品监督管理局(Food and Drug Administration, FDA)先后批准11项新适应症或新药,涵盖表皮生长因子受体(epidermal growth factor receptor, EGFR)、KRAS、间质-上皮细胞转化因子(mesenchymal-epithelial transition, MET)、间变性淋巴瘤激酶(anaplastic lymphoma kinase, ALK)及人表皮生长因子受体2(human epidermal growth factor receptor 2, HER2)等多个分子通路,深刻改变了临床实践格局。与此同时,中国创新药在多个靶点实现全球首创(first-in-class)突破,抗体药物偶联物(antibody-drug conjugate, ADC)技术快速迭代,微小残留病灶(minimal residual disease, MRD)指导的个体化治疗策略逐步成熟。本报告系统梳理上述创新靶点的关键临床试验数据与监管审批进展,分析竞争格局演变与技术趋势,为临床诊疗决策和研发投资布局提供循证参考。
主要发现概述
在EGFR通路,MARIPOSA III期研究确立埃万妥单抗(amivantamab)联合拉泽替尼(lazertinib)为EGFR突变晚期NSCLC一线治疗新标准,总生存期(overall survival, OS)风险比(hazard ratio, HR)为0.75。FLAURA-2研究将奥希替尼(osimertinib)联合化疗的中位OS推至47.5个月,刷新了该人群生存纪录。辅助领域,ADAURA研究显示奥希替尼术后辅助治疗5年OS率高达88%,LAURA研究则将其获益拓展至III期不可切除患者,中位无进展生存期(progression-free survival, PFS)达39.1个月。此外,舒沃替尼(sunvozertinib)获FDA批准,成为首款针对EGFR外显子20插入突变的国产靶向药。
在KRAS通路,6款KRAS G12C抑制剂已获FDA批准,竞争格局趋于饱和。值得关注的是,垂直抑制策略取得关键验证:KROCUS研究中埃万妥单抗联合索托拉西布(sotorasib)在初治患者中客观缓解率(objective response rate, ORR)达80%。针对更广泛的RAS突变,Zoldonrasib在KRAS G12D突变患者中ORR达61%,多款Pan-RAS抑制剂展现出覆盖全部RAS突变亚型的广谱抗肿瘤活性,其中RMC-6236在胰腺癌中OS HR低至0.40。
在MET通路,SACHI III期研究证实赛沃替尼(savolitinib)联合奥希替尼治疗MET扩增耐药患者的PFS HR为0.34,确立MET抑制剂联合EGFR-TKI为耐药后标准策略。Teliso-V成为首个获FDA批准的c-MET ADC药物,填补了该领域空白。目前已有5款MET酪氨酸激酶抑制剂(tyrosine kinase inhibitor, TKI)上市,MET通路治疗选择日益丰富。
在罕见驱动基因领域,CROWN研究5年随访数据显示洛拉替尼(lorlatinib)一线治疗ALK阳性患者的5年PFS率首次突破60%,ALINA研究显示阿来替尼(alectinib)辅助治疗的疾病无生存期(disease-free survival, DFS)HR低至0.24。Repotrectinib在ROS1阳性患者中展现出色疗效,中位PFS达35.7个月。
跨靶点层面,ADC药物分化明显:TROPION-Lung01验证组织因子靶向ADC的获益边界,而TROP2靶向ADC面临疗效与毒性平衡的挑战。免疫治疗领域,中国自主研发的依沃西单抗(ivonescimab)在头对头III期研究中击败帕博利珠单抗(pembrolizumab),PFS HR为0.51,标志着中国创新药从”跟跑”迈向”领跑”。2024年中国NSCLC领域共达成94项药物出海交易,总交易金额逾120亿美元。MRD指导的个体化治疗策略从临床试验走向真实世界应用,正在重塑NSCLC的全程管理模式。
关键词:非小细胞肺癌;靶向治疗;EGFR;KRAS G12C;MET;抗体药物偶联物;免疫治疗;个体化治疗
1. EGFR突变NSCLC靶向治疗进展
表皮生长因子受体(epidermal growth factor receptor, EGFR)突变是非小细胞肺癌(non-small cell lung cancer, NSCLC)中最常见的驱动基因变异,在亚洲人群肺腺癌中发生率约为40%-50%。自2004年首个EGFR酪氨酸激酶抑制剂(tyrosine kinase inhibitor, TKI)吉非替尼(Gefitinib)上市以来,EGFR靶向治疗历经一代可逆性TKI、二代不可逆性泛ErbB TKI、三代C797S靶向TKI的迭代演进,至2024-2025年,以Amivantamab联合Lazertinib为代表的”垂直抑制”策略在一线治疗中实现了对三代TKI单药的历史性超越,标志着EGFR突变NSCLC治疗格局进入联合治疗与个体化精准选择的新纪元。与此同时,奥希替尼(Osimertinib)辅助治疗在可切除NSCLC中的总生存获益、不可切除III期放化疗后巩固治疗的突破性无进展生存数据,以及首个新辅助三代TKI阳性III期结果的公布,共同勾勒出EGFR靶向治疗从晚期向全病程前移的完整图景。本章将从一线治疗格局重塑、早期与局部晚期适应症拓展、exon 20插入突变(exon 20 insertion, ex20ins)治疗突破、耐药克服策略、中国原研药物竞争格局及给药优化六个维度,系统梳理2024-2026年EGFR突变NSCLC靶向治疗领域的重大进展。
1.1 EGFR-TKI一线治疗格局重塑
1.1.1 MARIPOSA研究最终分析:Amivantamab联合Lazertinib的PFS和OS双获益
MARIPOSA研究是首个在一线治疗中以总生存期(overall survival, OS)为主要终点、头对头击败奥希替尼单药的III期随机对照试验,其最终结果发表于《新英格兰医学杂志》(NEJM 2025)。该研究共纳入1074例既往未经治疗的EGFR敏感突变(exon 19 deletion, ex19del或L858R)晚期NSCLC患者,按2:2:1比例随机分配至Amivantamab(EGFR/c-MET双特异性抗体)联合Lazertinib(三代EGFR-TKI)组、奥希替尼单药组或Lazertinib单药组(后两组分别为试验组和对照组)。
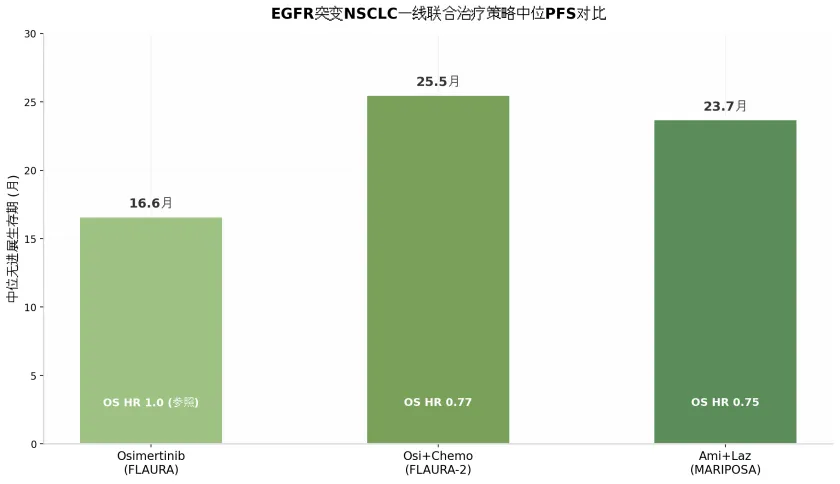
在主要终点方面,中位随访37.8个月时,Amivantamab联合Lazertinib组的中位无进展生存期(median progression-free survival, mPFS)达到23.7个月,较奥希替尼单药组的16.6个月显著延长(风险比 [hazard ratio, HR] 0.70,95%置信区间 [confidence interval, CI] 0.58-0.85;P<0.001)。联合组的中位OS尚未达到(not reached, NR;95%CI 42.9个月-NR),而奥希替尼组为36.7个月(HR 0.75,95%CI 0.61-0.92;P=0.005),死亡风险降低25%。两组3年OS率分别为60%和51%。这是自2018年FLAURA研究确立奥希替尼一线标准地位以来,首个在OS层面实现统计学显著性超越的联合方案。
值得注意的是,MARIPOSA研究在亚洲亚组中同样展现出一致性的OS获益(HR 0.75),客观缓解率(objective response rate, ORR)达86%,中位缓解持续时间(duration of response, DoR)为25.8个月,均优于奥希替尼单药组(ORR 85%,中位DoR 16.8个月)。基于该研究数据,美国食品药品监督管理局(Food and Drug Administration, FDA)于2024年8月批准Amivantamab联合Lazertinib用于EGFR敏感突变晚期NSCLC的一线治疗,美国国家综合癌症网络(National Comprehensive Cancer Network, NCCN)指南将其列为1类推荐。
然而,联合方案的毒性谱值得临床关注。Amivantamab联合Lazertinib组3级及以上不良事件(adverse event, AE)发生率为80%,显著高于奥希替尼单药组的52%,主要毒性包括皮肤相关事件(皮疹、甲沟炎、皮肤干燥)、静脉血栓栓塞(venous thromboembolism, VTE,发生率11%)和输注相关反应(infusion-related reaction, IRR,发生率63%)。COCOON和SKIPPirr两项II期研究表明,通过预防性口服和局部抗生素联合保湿剂的强化皮肤管理策略,可将2级及以上皮肤AE从75%降至42%(比值比 [odds ratio, OR] 0.24,P<0.0001);而预防性抗凝和分阶段输注策略则可显著降低VTE和IRR的发生率和严重程度。
1.1.2 FLAURA-2研究最终OS数据:化疗增效策略的循证依据
FLAURA-2研究探索了另一种一线联合策略——在奥希替尼基础上联合含铂双药化疗。该III期试验纳入557例初治EGFR敏感突变晚期NSCLC患者,随机分配至奥希替尼联合化疗(培美曲塞+卡铂/顺铂,4周期后培美曲塞维持)组或奥希替尼单药组。
2025年10月发表于NEJM的最终OS分析显示,联合组mOS为47.5个月,较单药组的37.6个月延长近10个月(HR 0.77,95%CI 0.61-0.96;P=0.02),死亡风险降低23%。3年OS率分别为63%和51%。此前2023年公布的主要分析显示,mPFS为25.5个月对比16.7个月(HR 0.62,P<0.001)。中位DoR方面,联合组为24.0个月,单药组为15.3个月。
FLAURA-2的亚组分析揭示,基线存在中枢神经系统(central nervous system, CNS)转移的患者从联合化疗中获益更为显著(CNS mOS 40.9对比29.7个月,HR 0.72),提示高肿瘤负荷和高转移风险人群可能是化疗增效策略的优先获益群体。该方案于2024年2月获FDA批准一线适应症。然而,联合化疗的毒性代价不容忽视:3级及以上AE发生率为70%,对比单药组的34%,主要包括骨髓抑制、恶心和疲劳;奥希替尼停药率分别为12%和7%。这一毒性特征与MARIPOSA方案的免疫-皮肤毒性谱形成鲜明差异,为临床决策提供了不同的风险-获益权衡依据。
一线联合治疗策略PFS对比
1.1.3 Amivantamab+Lazertinib vs Osimertinib+化疗的间接比较
MARIPOSA和FLAURA-2两项III期试验虽均以奥希替尼单药为共同对照,但并未进行头对头直接比较。在临床实践中,两种联合策略的选择需基于患者个体特征、合并症和耐受性进行综合判断。以下从关键疗效和安全性维度进行间接比较分析。
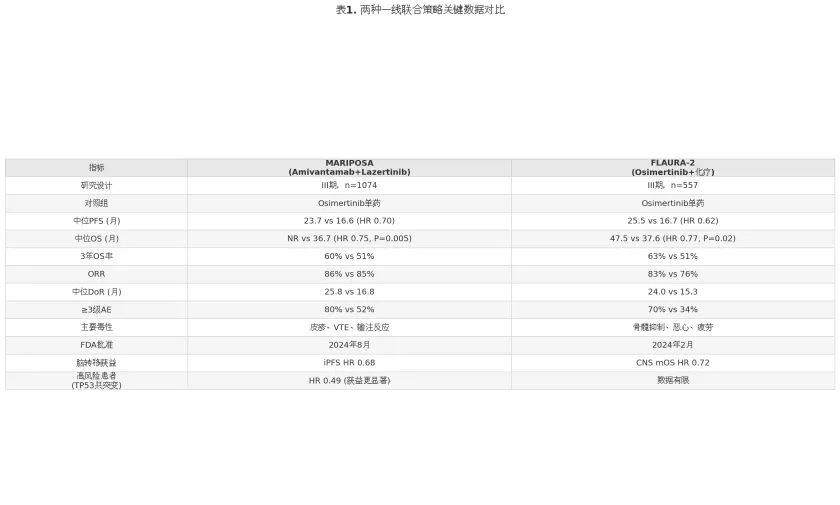
表1
上述数据表明,两种联合策略在OS层面获益幅度相近(HR 0.75 vs 0.77),PFS获益亦处于同一数量级。但毒性谱差异显著:MARIPOSA方案以皮肤毒性、VTE和IRR为主要挑战,FLAURA-2方案则以骨髓抑制和消化道毒性更为突出。在临床决策中,对于合并脑转移、TP53共突变或肝转移的高风险患者,MARIPOSA方案可能提供更优的获益——MARIPOSA研究亚组分析显示,TP53共突变患者接受Amivantamab联合Lazertinib的OS获益更为显著(HR 0.49),可能与该方案同时阻断EGFR和MET通路、延缓旁路耐药激活有关。而对于体能状态良好、肿瘤负荷较高且对化疗耐受性较好的患者,FLAURA-2方案提供了略长的mPFS数值(25.5个月 vs 23.7个月)和成熟的OS数据(mOS 47.5个月)。
需要指出的是,间接比较存在固有局限性:两项试验的入组人群基线特征、随访时间和交叉治疗比例存在差异,无法排除人群异质性对比较结果的干扰。因此,目前尚不能得出两种联合策略的绝对优劣排序。正在进行的真实世界研究和网状Meta分析有望为这一临床决策困境提供更多证据支持。对于标准风险患者,奥希替尼单药仍是性价比合理的可选项,尤其在对联合治疗毒性顾虑较大或医疗资源受限的临床场景中。
1.1.4 MARIPOSA研究揭示的耐药优势:“垂直抑制”延缓耐药
MARIPOSA研究另一项具有深远意义的发现来自治疗后获得性耐药机制的深入分析(ESMO 2024,LBA55)。研究者对两组进展患者的循环肿瘤DNA(circulating tumor DNA, ctDNA)和组织样本进行了全面的基因组学分析,结果揭示了两种治疗策略在耐药谱上的显著差异。
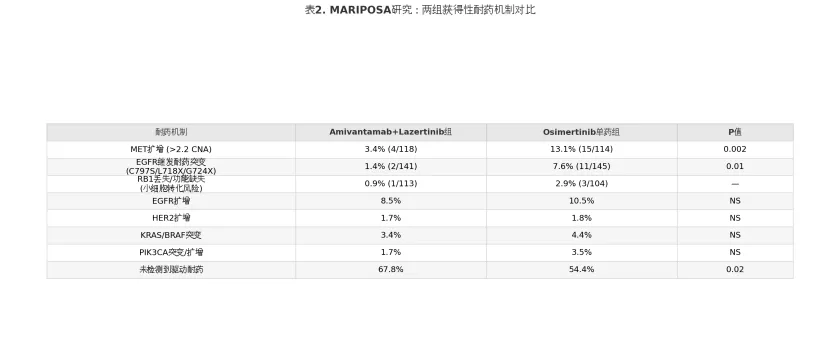
表2
这一数据从机制层面验证了”垂直抑制”(vertical inhibition)策略的合理性:Amivantamab作为EGFR/c-MET双特异性抗体,同时阻断受体酪氨酸激酶(receptor tyrosine kinase, RTK)层面的EGFR信号和旁路MET通路的激活,从而显著降低了MET扩增驱动的获得性耐药发生率(3.4% vs 13.1%,P=0.002)。EGFR继发耐药突变(包括C797S、L718X和G724X等)在联合组的发生率同样大幅降低(1.4% vs 7.6%,P=0.01),可能归因于Lazertinib对EGFR通路的持续抑制与Amivantamab的EGFR胞外域阻断的协同效应。此外,RB1丢失(与小细胞肺癌 [small cell lung cancer, SCLC] 转化相关)在联合组亦呈减少趋势(0.9% vs 2.9%)。
这一发现具有重要的临床转化价值。MET扩增是奥希替尼一线治疗后最常见的获得性旁路耐药机制(发生率约15%),传统上需在耐药后检测确认,再启用MET抑制剂联合治疗。MARIPOSA研究提示,一线即采用EGFR/MET双靶阻断可能从根本上改变耐药发生的时间轨迹和分子谱,使更多患者在进展时进入”无明确驱动耐药”状态(67.8% vs 54.4%),从而为后续以抗体偶联药物(antibody-drug conjugate, ADC)或化疗为基础的泛靶治疗创造更有利的条件。
1.2 奥希替尼适应症向早期和局部晚期拓展
1.2.1 ADAURA研究5年OS数据:术后辅助标准治疗的确立
ADAURA研究是改变可切除EGFR突变NSCLC辅助治疗格局的里程碑式III期试验。该研究纳入682例IB期(肿瘤直径≥4cm)、II期和IIIA期(根据美国癌症联合委员会 [American Joint Committee on Cancer, AJCC] 第7版分期)完全切除的EGFR敏感突变NSCLC患者,按1:1随机分配至奥希替尼(80mg每日一次,口服3年)辅助组或安慰剂组。
2023年7月发表于NEJM的最终OS分析(中位随访约60个月)显示,在II-IIIA期患者中,奥希替尼组5年OS率为85%,显著高于安慰剂组的73%(HR 0.49,95.03%CI 0.33-0.73;P<0.001)。IB-IIIA期整体人群的5年OS率分别为88%和78%(HR 0.49),死亡风险降低51%。无病生存期(disease-free survival, DFS)方面,II-IIIA期患者的中位DFS为65.8个月对比21.9个月(HR 0.23)。安慰剂组54%的患者在复发后接受了含奥希替尼的后续治疗,而奥希替尼组仅22%需要后续治疗,提示辅助奥希替尼已将大部分复发事件推迟或消除。
ADAURA研究是首个证实辅助EGFR-TKI可显著改善OS的III期随机对照试验,彻底改变了此前以辅助化疗为主的治疗范式。2024年更新的NCCN指南将奥希替尼辅助推荐类别从2A类提升至1类。2025年ASCO年会公布的MRD(分子残留病灶,molecular residual disease)分析进一步表明,辅助奥希替尼治疗期间的MRD动态监测可提前约4.7个月预警复发,且MRD阳性患者从辅助靶向治疗中的获益更为显著。多项基于MRD的个体化辅助治疗策略探索性试验(包括CTONG 2201等7项研究)正在进行中,有望在未来3-5年内推动辅助治疗从”固定疗程”模式向”MRD指导的动态调整”模式演进。
1.2.2 LAURA研究:III期不可切除EGFR突变NSCLC的突破性进展
对于不可切除的III期EGFR突变NSCLC,同步放化疗(concurrent chemoradiotherapy, cCRT)后免疫巩固治疗(度伐利尤单抗 [Durvalumab])是驱动基因阴性患者的标准治疗,但EGFR突变患者对免疫治疗的应答率较低,此前缺乏针对性的巩固治疗策略。LAURA III期试验填补了这一证据空白。
LAURA研究纳入143例接受cCRT后未出现疾病进展的III期不可切除EGFR突变NSCLC患者,2:1随机分配至奥希替尼(80mg每日一次)巩固治疗组或安慰剂组,直至疾病进展或最长治疗3年。2024年ASCO年会首次公布的结果显示,奥希替尼组mPFS达到39.1个月,而安慰剂组仅为5.6个月(HR 0.16,95%CI 0.10-0.24;P<0.001),进展风险降低84%。2025年更新的OS数据继续显示奥希替尼相比安慰剂的获益趋势,二次无进展生存期(progression-free survival after next line of therapy, PFS2)亦得到改善。
LAURA研究的意义不仅在于其卓越的PFS数据,更在于确立了EGFR突变检测在III期不可切除NSCLC患者中的必要性——既往III期NSCLC的分子检测率远低于IV期患者,LAURA研究结果的公布将有力推动该人群常规EGFR检测的实施。FDA于2024年9月批准奥希替尼用于经cCRT后疾病未进展的不可切除III期EGFR突变NSCLC的巩固治疗,NCCN指南将其列为1类推荐。该适应症的获批进一步拓展了奥希替尼在NSCLC治疗全病程中的覆盖范围,使其成为横跨辅助(ADAURA)、局部晚期巩固(LAURA)和晚期一线(FLAURA/FLAURA-2)三个关键治疗阶段的”全程覆盖”型靶向药物。
1.2.3 NeoADAURA研究:首个新辅助三代EGFR-TKI III期阳性结果
新辅助靶向治疗是可切除NSCLC领域的研究前沿。NeoADAURA III期试验评估了奥希替尼或奥希替尼联合化疗作为新辅助治疗在可切除II-IIIB期(AJCC第8版)EGFR突变NSCLC中的疗效。
该研究将患者随机分配至三个治疗组:奥希替尼单药新辅助组、奥希替尼联合化疗组和单纯化疗组,新辅助治疗后接受手术切除和辅助奥希替尼(在奥希替尼组)或辅助化疗(在化疗组)。主要终点为病理完全缓解率(pathological complete response, pCR)和主要病理缓解率(major pathological response, MPR)。2024年公布的结果显示,奥希替尼单药组的MPR达到25%-26%,奥希替尼联合化疗组的MPR更高,而单纯化疗组仅为2%。12个月无事件生存率(event-free survival, EFS)在奥希替尼组达93%-95%,显著优于化疗组。
NeoADAURA研究是首个报道新辅助三代EGFR-TKI III期阳性结果的研究,证实了新辅助靶向治疗在EGFR突变可切除NSCLC中的可行性。尽管MPR数值(25%-26%)低于新辅助免疫治疗在驱动基因阴性NSCLC中的报道(约36%-37%,CheckMate 816研究),但考虑到化疗对照组MPR仅2%,奥希替尼带来的绝对获益幅度(约23个百分点)仍具有重要临床意义。新辅助奥希替尼的安全性特征良好,未显著增加手术并发症发生率,这为其在特定患者群体中的临床转化奠定了基础。NeoADAURA研究已于2025年提交中国CDE新药上市申请(new drug application, NDA),预计2026-2027年有望获批,届时将进一步完善奥希替尼在NSCLC全病程中的治疗拼图。
1.2.4 辅助与新辅助靶向治疗的临床路径整合
奥希替尼已在EGFR突变NSCLC的多个治疗阶段取得阳性III期数据,形成了从可切除早期到不可切除局部晚期、再到晚期转移性疾病的全病程覆盖。
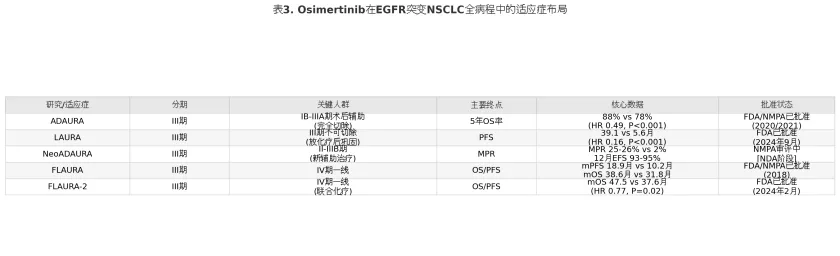
表3
上述数据综合呈现了奥希替尼在EGFR突变NSCLC各治疗阶段的布局。值得注意的是,不同治疗阶段的风险比(HR)反映了疾病背景对靶向治疗敏感性的影响:III期不可切除(LAURA)的PFS获益最为显著(HR 0.16),可能与放化疗后肿瘤负荷极低、奥希替尼在微小残留病灶(minimal residual disease, MR)状态下的高效清除能力有关;而IV期一线治疗(FLAURA)的OS获益相对有限(HR 0.79),提示晚期肿瘤的高度异质性和多重耐药机制对单药靶向治疗构成了根本性挑战,这也为前述联合治疗策略(MARIPOSA、FLAURA-2)的临床价值提供了背景支撑。
奥希替尼多阶段应用疗效数据
在临床路径整合方面,目前尚需解决的关键问题包括:第一,辅助与新辅助靶向治疗的序贯策略——对于降期后接受手术的患者,新辅助奥希替尼后的最佳辅助治疗时长仍有待探索;第二,MRD指导下的个体化治疗强度调整——部分MRD阴性低复发风险患者可能无需接受完整的3年辅助治疗,而MRD持续阳性患者则可能需要延长治疗或联合其他治疗手段;第三,耐药后的精准再治疗——辅助或新辅助奥希替尼进展后的耐药机制与治疗选择正在多项前瞻性研究(包括ORCHARD平台试验等)中进行探索。上述方向的研究成果预计将在2026-2028年陆续公布,有望推动EGFR突变NSCLC从”精准诊断+固定方案”模式向”动态监测+自适应调整”模式转型。
1.3 EGFR exon 20插入突变治疗突破
1.3.1 PAPILLON研究:Amivantamab联合化疗一线标准的确立
EGFR ex20ins突变约占所有EGFR突变的4%-12%,因突变位置和插入序列的高度异质性,对传统的EGFR-TKI(包括奥希替尼)应答有限,中位PFS仅约2-4个月,曾是EGFR突变NSCLC中最难治的亚型之一。PAPILLON III期试验首次在一线治疗中证实,Amivantamab联合化疗可显著改善ex20ins突变患者的预后。
PAPILLON研究纳入308例既往未经治疗的EGFR ex20ins晚期NSCLC患者,1:1随机分配至Amivantamab联合化疗(卡铂+培美曲塞)组或单纯化疗组。2023年10月发表于NEJM的结果显示,联合组mPFS为11.4个月,显著优于化疗组的6.7个月(HR 0.40,95%CI 0.30-0.53;P<0.001),进展风险降低60%。ORR分别为73%和47%。OS数据尚不成熟,但已显示获益趋势(HR 0.67,成熟度33%)。CNS活性方面,联合组的颅内ORR(intracranial ORR, iORR)达67%。
PAPILLON研究确立了Amivantamab联合化疗作为EGFR ex20ins一线治疗的标准方案。FDA于2024年3月批准该适应症,NCCN指南将其列为1类推荐。该方案的毒性谱与MARIPOSA研究一致,以皮肤相关事件、VTE和IRR为主要关注点,COCOON研究证实的强化皮肤管理策略同样适用于该人群。值得注意的是,Amivantamab联合化疗方案需要每2周一次的静脉输注,患者治疗负担相对较重,这为口服ex20ins抑制剂的一线应用探索预留了空间。
1.3.2 舒沃替尼(Sunvozertinib):中国首个独立研发在美获批的First-in-class肺癌新药
舒沃替尼(Sunvozertinib,研发代号DZD9008)是由迪哲医药自主研发的高选择性、不可逆EGFR ex20ins抑制剂。2025年7月2日,FDA基于WU-KONG1B II期研究结果,加速批准舒沃替尼(商品名Zegfrovy)用于治疗含铂化疗后进展的EGFR ex20ins突变局部晚期或转移性NSCLC。这是两项具有标志性意义的里程碑:其一,舒沃替尼成为全球首个且唯一在美国获批的EGFR ex20ins口服靶向药物;其二,这也是中国首个独立研发、在美获批的First-in-class(全球首创)肺癌新药。
WU-KONG1B研究纳入经治EGFR ex20ins突变NSCLC患者,关键队列(200mg每日一次剂量组)的确认ORR为46%(95%CI 35%-57%),中位DoR为11.1个月(95%CI 8.2个月-NR)。脑转移患者的颅内ORR达48.4%,展现了良好的中枢活性。剂量探索分析显示,300mg组ORR略升至47.2%,且在脑转移患者中iORR进一步提高至52.4%(对比200mg组的28.6%),提示高剂量可能对CNS转移患者更有利;但FDA批准的推荐剂量仍为200mg每日一次,基于该剂量的获益-风险评估最为均衡。
舒沃替尼在中国的研发进程更为迅速——该药已于2023年8月获中国国家药品监督管理局(National Medical Products Administration, NMPA)批准,早于美国批准近两年。WU-KONG28 III期试验(舒沃替尼对比化疗一线治疗ex20ins NSCLC)已完成入组,覆盖16个国家和地区,预计2026年公布数据。若一线适应症获批,舒沃替尼将有望与Amivantamab联合化疗形成”口服vs静脉”的竞争格局,为临床医生和患者提供更多选择。此外,舒沃替尼联合安罗替尼(多靶点抗血管TKI)一线治疗的早期数据显示ORR超80%、疾病控制率(disease control rate, DCR)达100%,为无化疗方案的探索开辟了新方向。
1.3.3 Zipalertinib、Furmonertinib等其他ex20ins抑制剂进展
在舒沃替尼之外,多款ex20ins靶向药物正处于不同的临床开发阶段,形成了日益丰富的治疗选择矩阵。
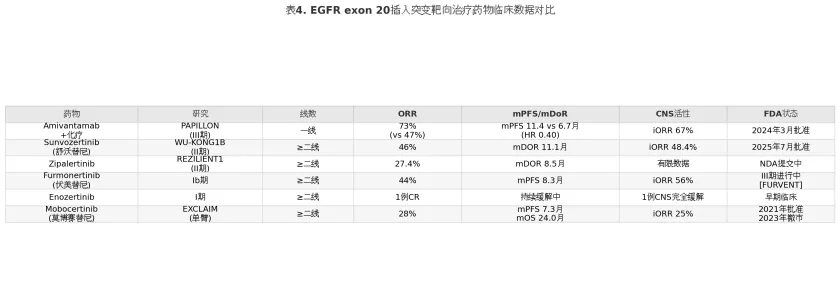
表4
Zipalertinib(CLN-081/TAS-6417)是由Cullinan Oncology/再鼎医药开发的高选择性不可逆EGFR抑制剂,在REZILIENT1 II期研究中,经治ex20ins NSCLC患者(含Amivantamab经治人群)的确认ORR为27.4%,中位DoR 8.5个月。该药的差异化特征在于对野生型EGFR的较低亲和力,可能带来更优的皮肤和消化道耐受性。REZILIENT3 III期试验(Zipalertinib联合化疗对比化疗一线治疗ex20ins NSCLC)正在进行中,预计2026年上半年完成入组,若结果阳性将为ex20ins一线治疗增加另一口服选择。
Furmonertinib(伏美替尼,AST2818)作为已在中国获批的经典EGFR敏感突变三代TKI,在ex20ins领域亦展现了临床活性。Ib期研究中,高剂量Furmonertinib在ex20ins患者中的ORR为44%,mPFS为8.3个月,颅内ORR达56%。FURVENT全球III期试验(FURMO-004,Furmonertinib对比化疗一线治疗ex20ins NSCLC)已于2025年第一季度完成入组,预计2026年中数据读出。若获FDA批准,伏美替尼将成为继舒沃替尼之后第二个在美获批的ex20ins口服靶向药。该药已通过FDA突破性疗法认定,由ArriVent BioPharma负责全球商业化。
Enozertinib(ORIC-114)是一款强调高脑渗透性的ex20ins抑制剂,在临床前研究中展现了优异的血脑屏障(blood-brain barrier, BBB)穿透能力。I期临床试验中,1例ex20ins伴脑转移患者实现了所有病灶(包括CNS病灶)的完全缓解(complete response, CR),这一病例虽样本量极小,但为脑转移为主的ex20ins患者群体提供了概念验证(proof-of-concept)证据。
对比来看,ex20ins治疗领域已呈现”双轨并行”格局:以Amivantamab联合化疗为代表的”大分子联合化疗”策略在一线治疗中数据最为成熟(PFS HR 0.40),但需静脉给药且毒性管理复杂;以舒沃替尼、Zipalertinib和Furmonertinib为代表的”口服小分子单药”策略则在便利性和经治人群覆盖方面具有优势。未来一线治疗的选择将在疗效、便利性、耐受性和成本之间进行综合权衡。
1.3.4 ex20ins突变检测标准化:DNA-NGS漏检率与RNA-NGS金标准
ex20ins突变的高度异质性(迄今已报道超过100种不同的插入变异)对临床检测提出了特殊挑战。基于DNA的新一代测序技术(next-generation sequencing, NGS)在ex20ins检测中存在固有的技术局限:由于20号外显子区域的GC含量较高且插入序列长度变化大,DNA-NGS的探针捕获和比对算法可能无法完全覆盖所有变异类型,导致漏检率约达50%。多项研究表明,RNA-NGS因直接检测转录本水平的融合和插入事件,可有效规避DNA层面的覆盖盲区,成为ex20ins检测的”金标准”。
在临床实践中,对于EGFR突变检测结果阴性但临床特征(不吸烟、女性、腺癌)高度提示EGFR驱动可能的患者,建议采用RNA-NGS进行复核。此外,液体活检(ctDNA-based NGS)在ex20ins检测中的敏感性较组织样本进一步降低,阴性结果不能排除ex20ins突变存在。伴随舒沃替尼等ex20ins特异性TKI的获批,检测标准化和检测率的提升将成为确保更多患者获益于精准治疗的关键基础设施。
1.4 四代EGFR-TKI与耐药克服策略
1.4.1 C797S突变发生率与临床特征
尽管三代EGFR-TKI显著改善了EGFR突变NSCLC患者的预后,获得性耐药仍是制约长期疗效的核心瓶颈。EGFR C797S突变是奥希替尼治疗后最常见的EGFR依赖性耐药机制,其发生率因治疗线数而异:一线奥希替尼治疗后发生率约7%(FLAURA研究,基于ctDNA检测91例耐药患者),二线奥希替尼(T790M阳性经一代/二代TKI治疗后)发生率升至14%-18%,三线及后线治疗中最高可达22%-29%。C797S突变发生在奥希替尼与EGFR蛋白结合的ATP口袋区域,通过空间位阻效应阻止药物结合。
C797S突变的临床意义在于其与共存突变的构象关系:当C797S与T790M呈反式(trans)构象时,可采用第一代联合第三代TKI(如吉非替尼+奥希替尼)进行克服;当呈顺式(cis)构象时,现有EGFR-TKI均无效,推荐布格替尼(Brigatinib,ALK抑制剂)联合西妥昔单抗(Cetuximab,EGFR单克隆抗体)方案(基于小样本回顾性数据,ORR约30%);若C797S为单独出现(无T790M),则可换用一代或二代EGFR-TKI。除C797S外,其他罕见EGFR继发突变(L718Q、L792X、G724S等)总发生率不足5%,目前缺乏有效的靶向治疗策略。
从耐药机制全景来看,MET扩增是奥希替尼一线治疗后最常见的旁路耐药机制(发生率约15%,基于FLAURA研究ctDNA数据),其次是EGFR扩增(9%-19%)、PIK3CA突变/扩增(6%-7%)、HER2扩增(2%)和BRAF V600E突变(3%)。组织学转化(包括SCLC转化,发生率2%-4%;鳞状细胞转化,罕见)和上皮-间质转化(epithelial-mesenchymal transition, EMT)是不依赖于特定基因突变的表型耐药机制,需通过组织再活检确诊。
1.4.2 四代TKI研发管线:前景与挑战并存
针对C797S突变及复合耐药(如L858R/T790M/C797S三重突变),四代EGFR-TKI的研发已推进多年,但进展坎坷,反映出该领域的固有挑战。
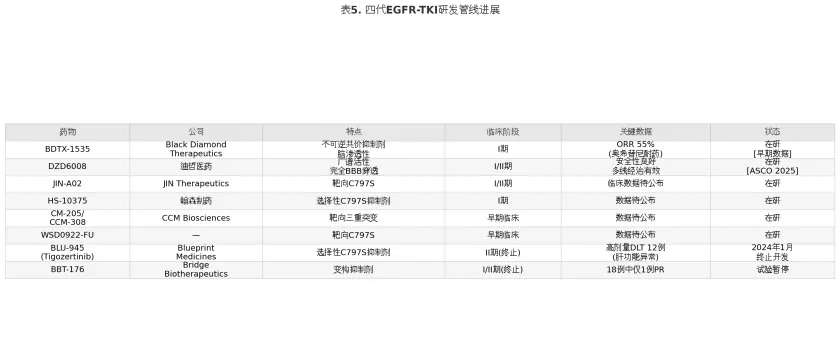
表5
BLU-945(Tigozertinib)曾是最受关注的四代TKI候选药物之一,其高选择性靶向C797S的设计理念在体外研究中表现优异。然而,2024年1月Blueprint Medicines宣布终止BLU-945的开发——在II期研究中,高剂量组(400-600mg/日)出现12例剂量限制性毒性(dose-limiting toxicity, DLT),主要为肝功能异常,治疗窗狭窄导致无法达到有效血药浓度。BBT-176(Bridge Biotherapeutics)的变构抑制剂策略同样遭遇挫折,18例患者中仅1例达到部分缓解(partial response, PR),试验已被暂停。
在研管线中,BDTX-1535(Black Diamond Therapeutics)是目前临床数据最有前景的四代TKI候选。该药为不可逆共价EGFR抑制剂,具有良好脑渗透性,2024年美国癌症研究协会(American Association for Cancer Research, AACR)年会公布的早期数据显示,在奥希替尼耐药患者中ORR达55%,且低剂量组未观察到DLT。DZD6008(迪哲医药)作为另一款具有完全血脑屏障穿透能力的四代TKI,在2025年ASCO年会报道了I/II期初步数据,在重度预治疗(含奥希替尼耐药及多线治疗失败)患者中展现了良好的安全性和抗肿瘤活性。
从商业化和临床需求角度审视,四代TKI面临的核心困境在于目标人群规模有限:C797S突变在一线奥希替尼后发生率仅约7%,且MARIPOSA研究已证实Amivantamab联合Lazertinib可将EGFR继发耐药突变率从7.6%降至1.4%。若联合方案成为一线标准,C797S突变作为耐药靶点的临床需求将进一步缩小。因此,四代TKI的商业前景高度依赖于能否拓展至更广泛的耐药场景(如同时覆盖MET扩增、HER2扩增等多重耐药机制),或在联合治疗中发挥增效作用。
1.4.3 双靶联合克服耐药:Amivantamab+化疗与ADC”通用解”
对于奥希替尼耐药后的治疗选择,当前循证证据最充分的方案来自MARIPOSA-2 III期试验。该研究纳入奥希替尼耐药后的EGFR突变晚期NSCLC患者,随机分配至Amivantamab+化疗(卡铂+培美曲塞)组或单纯化疗组。结果显示,联合组mPFS为6.3个月(研究者评估)或8.3个月(盲法独立中心评估),对比化疗组的4.2个月(HR 0.48);OS为17.7个月对比15.3个月(HR 0.73);PFS2(接受后续治疗后的PFS)为16.0个月对比11.6个月(HR 0.64,P=0.002)。该方案已获NCCN指南1类推荐,成为奥希替尼耐药后的标准治疗选择之一。
针对特定耐药机制的双靶联合策略亦取得重要进展。MET扩增耐药方面,SACHI中国III期试验证实,赛沃替尼(Savolitinib,选择性MET抑制剂)联合奥希替尼对比化疗在MET扩增耐药患者中mPFS为8.2个月对比4.5个月(HR 0.34,P<0.0001),ORR为58%对比34%。该方案于2025年6月获中国NMPA批准。全球III期SAFFRON试验已完成入组,评估该方案在全球人群中的疗效,并已获FDA快速通道资格。
抗体偶联药物(ADC)正成为耐药后治疗的重要”通用解”(universal solution)——即不依赖于特定耐药机制的广谱治疗选择。Dato-DXd(Datopotamab deruxtecan,TROP2 ADC)基于TROPION-Lung05 II期研究数据,于2025年6月获FDA加速批准用于EGFR突变NSCLC后线治疗。在该研究中,EGFR突变亚组(n=117)的ORR为43.6%,mPFS为5.8个月,mOS为15.6个月。值得注意的是,Dato-DXd在EGFR突变经治人群中的疗效优于非选择性NSCLC人群(TROPION-Lung01全人群OS未达终点),提示驱动基因阳性肿瘤可能对ADC具有特殊的敏感性,其机制可能与更高的TROP2表达水平或更依赖增殖信号的特性有关,尚需基础研究验证。另一款HER3-DXd(Patritumab deruxtecan)在HERTHENA-Lung01 II期研究中ORR为29.8%,但HERTHENA-Lung02 III期试验因OS未达统计学显著性(16.0对比15.9个月),生物制品许可申请(biologics license application, BLA)于2025年5月被主动撤回。这一失败案例提示,ADC的临床成功不仅依赖于靶点表达,更取决于内化效率、载荷敏感性和患者筛选标志物的精准度。
1.5 中国原研三代EGFR-TKI全景
1.5.1 8款已获批国产三代TKI竞争格局
截至2025年底,中国市场已有8款三代EGFR-TKI获批上市(含进口奥希替尼和韩国合作开发的Lazertinib),形成了全球最密集的三代TKI竞争格局。其中,6款为中国原研药物,均通过与一代TKI(吉非替尼或埃克替尼)的头对头III期试验获得一线治疗适应症。
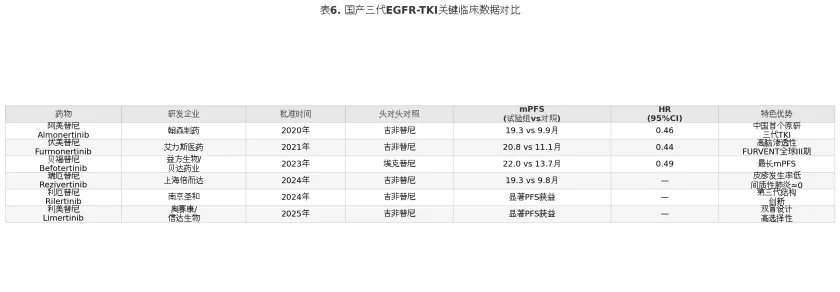
表6
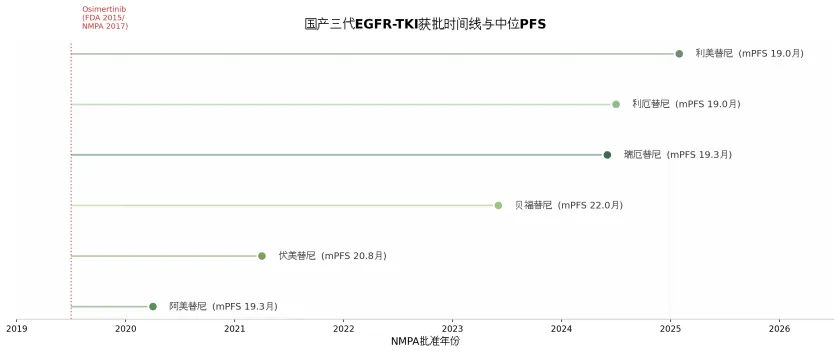
国产三代EGFR-TKI获批时间线
上述数据表明,国产三代TKI在与一代TKI的头对头比较中,PFS的HR值普遍介于0.44-0.49之间,mPFS介于19.3-22.0个月,获益幅度处于同一数量级。值得注意的是,这些试验均以一代TKI为对照而非奥希替尼,因此无法直接比较各药物之间的优劣。从差异化特征来看,伏美替尼以高脑渗透性为设计重点,在CNS病灶控制方面表现突出;贝福替尼在mPFS数值上达到22.0个月(对比埃克替尼13.7个月),为国产TKI中最高;瑞厄替尼则以安全性优势著称,间质性肺炎发生率接近零,皮疹发生率亦显著低于其他三代TKI,可能与其代谢产物对旁路信号通路无活性的药理学特征有关。
在中国市场,三代TKI的价格竞争已进入白热化阶段。2024年国家医保目录更新后,奥希替尼医保支付价格降至5580元/盒(降幅约64%),阿美替尼3520元/盒,伏美替尼3304元/盒(首年医保降幅79%)。估算年治疗费用(按医保报销前计算),奥希替尼约6.7万元,阿美替尼约4.2万元,伏美替尼约4.0万元。激烈的价格竞争和医保续约压力使得后上市产品的市场空间被严重压缩,2024-2025年获批的瑞厄替尼、利厄替尼和利美替尼面临的市场准入挑战尤为严峻。
1.5.2 TY-9591:全球首个头对头击败Osimertinib的EGFR-TKI
TY-9591是由同源康医药自主研发的第三代EGFR-TKI,其最具差异化的研发策略在于专注EGFR突变NSCLC脑转移这一高度未满足需求的临床场景。NSCLC脑转移发生率约20%-40%,EGFR突变患者的CNS转移风险更高,现有三代TKI(包括奥希替尼)的颅内客观缓解率约50%-70%,但中位颅内PFS仍有较大提升空间。
2025年ESMO Asia年会公布的LBA5摘要显示,TY-9591在随机III期试验中头对头击败了奥希替尼,用于初治EGFR突变晚期NSCLC伴CNS转移患者的一线治疗。该研究共入组224例患者,主要终点颅内ORR(iORR)显示TY-9591较奥希替尼有统计学显著性改善(具体数值待全文发表)。基于该阳性结果,TY-9591于2026年1月获中国CDE纳入优先审评程序,成为全球首个在头对头III期研究中证明优于奥希替尼的EGFR-TKI(尽管仅限于脑转移亚群)。
TY-9591的商业化前景高度依赖于两个因素:第一,III期试验完整数据的发表和审评结果——若iORR的绝对改善幅度达到20个百分点以上,将为临床提供具有实质性意义的替代选择;第二,L858R突变亚群中的疗效数据——该突变亚型对现有三代TKI的敏感性低于ex19del,且CNS转移发生率更高,若TY-9591在L858R人群中展现出更显著的差异化优势,将进一步巩固其市场定位。同源康医药计划推进TY-9591的中美双报,但在美国上市需补充国际多中心III期数据。
1.5.3 国产三代TKI出海困境与突破
中国三代EGFR-TKI虽已在国内市场形成激烈竞争,但国际化进程参差不齐,整体面临”国内拥挤、海外缺席”的结构性困境。
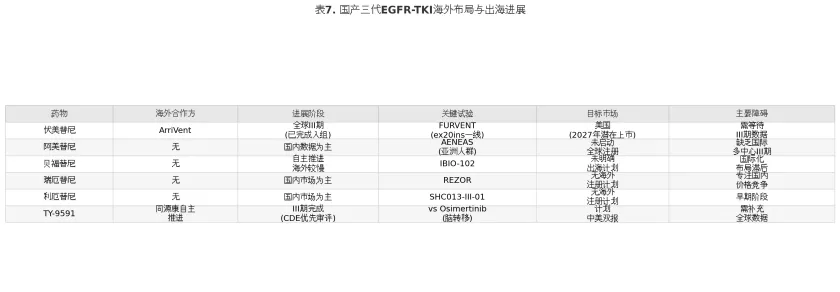
表7
国产三代TKI出海面临的核心障碍包括三个方面。其一,临床设计的局限性——所有国产三代TKI的注册试验均以一代TKI为对照,而非当前全球标准治疗奥希替尼。FDA和欧洲药品管理局(European Medicines Agency, EMA)在审评时,通常要求新药与所在适应症的最佳可用治疗(best available therapy)进行头对头比较,这使得基于”vs 吉非替尼/埃克替尼”数据的国产TKI在监管审评中处于不利地位。其二,人群代表性的局限——注册试验主要在中国人群中开展,缺乏足够的人种多样性和全球多中心数据,难以满足FDA对种族差异评估的要求。其三,商业化能力的短板——除伏美替尼通过与ArriVent的合作模式引入海外商业化经验外,多数中国药企缺乏自主的海外注册、市场准入和商业推广能力。
伏美替尼的国际化路径为国产TKI出海提供了可参考的范本。通过与ArriVent BioPharma的深度合作(保留股权+里程碑付款模式),伏美替尼已推进FURVENT全球III期试验(ex20ins一线适应症),于2025年Q1完成入组,预计2026年中数据读出,2027年美国潜在上市。海外里程碑付款总额超过7.65亿美元,体现了国际市场对差异化三代TKI的价值认可。恩沙替尼(Ensartinib,贝达药业ALK-TKI)通过自主开展覆盖21个国家的全球多中心III期试验,于2024年12月获FDA批准,为肺癌靶向药物的全链条自主出海模式提供了先例。
从全球竞争格局审视,中国三代TKI出海窗口正在收窄——奥希替尼专利保护期至2027-2030年(分区域),且MARIPOSA方案已在一线治疗中建立起新的疗效标杆。后上市的国产三代TKI若无明确的差异化优势(如显著优于奥希替尼的CNS活性、独特的安全性特征或创新的联合治疗策略),在美国和欧洲等成熟市场将面临严峻的定价和报销压力。因此,“差异化定位+国际合作+真实世界证据”的组合策略,可能成为中国三代TKI全球化突破的可行路径。
1.6 Amivantamab皮下制剂与给药优化
1.6.1 PALOMA-3研究:皮下制剂的非劣效与潜在优势
Amivantamab的静脉输注方案因输注时间长(首剂分两天给药,总计约6.5小时)和较高的IRR发生率(67%),在临床实践中对患者的治疗体验和医疗资源占用构成显著负担。PALOMA-3 III期试验评估了重组人透明质酸酶(rHuPH20)辅助的皮下注射(subcutaneous, SC)制剂在药代动力学、疗效和安全性方面是否非劣效于静脉输注(intravenous, IV)制剂。
PALOMA-3研究纳入418例奥希替尼耐药的EGFR突变晚期NSCLC患者,随机分配至SC Amivantamab+Lazertinib组或IV Amivantamab+Lazertinib组,联合化疗(卡铂+培美曲塞)。研究主要终点为第2周期第1天前的谷浓度(Ctrough)非劣效性。结果显示,SC制剂的Ctrough比值达到预设的非劣效界值,主要终点达成。更引人注目的是,SC组在多个关键疗效终点上显示出优于IV组的趋势:SC组mPFS为6.9个月对比IV组的5.8个月(HR 0.59),OS HR为0.62(95%CI 0.42-0.92;P=0.02),提示SC制剂可能带来生存获益。这一看似违反直觉的结果(SC优于IV)可能与SC给药的药代动力学特征有关——皮下注射后的药物吸收更为平缓持久,可能维持了更稳定的血药浓度和持续的受体占位。
在安全性方面,SC组的优势更为明显:IRR发生率从67%(IV组)骤降至16%(SC组),首剂中位椅位时间从6.5小时缩短至约7分钟(中位0.4小时)。3级及以上AE整体发生率SC组亦更低。患者偏好调查中,85%的患者明确表示偏好SC给药方式。
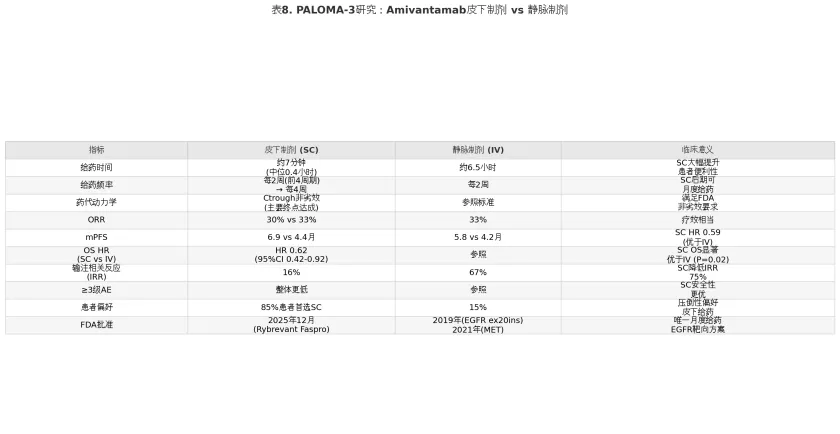
表8
1.6.2 2025年12月FDA批准皮下制剂:COCOON研究验证强化管理策略
FDA于2025年12月正式批准Amivantamab皮下制剂(商品名Rybrevant Faspro),用于既往接受过治疗的EGFR敏感突变和ex20ins晚期NSCLC患者。2026年2月,FDA进一步批准每4周给药方案(前4周期每2周给药,之后每4周给药),使Amivantamab成为目前唯一可月度给药的EGFR靶向治疗方案,患者便利性大幅提升。欧盟药品管理局(European Medicines Agency, EMA)人用医药产品委员会(Committee for Medicinal Products for Human Use, CHMP)已给予积极推荐。
在毒性管理方面,COCOON II期试验为临床实践提供了关键循证依据。该研究比较了强化皮肤管理(预防性口服多西环素/米诺环素+局部糖皮质激素+保湿剂)与标准管理在Amivantamab联合Lazertinib治疗患者中的效果。结果显示,强化管理组2级及以上皮肤AE发生率为42%,对比标准管理组的75%(OR 0.24,95%CI 0.13-0.45;P<0.0001),降幅达50%。强化管理方案未增加额外的安全性负担,患者生活质量评分(Patient-Reported Outcomes, PRO)亦有改善。这一策略已成为Amivantamab相关皮肤毒性的标准管理模式,并写入了NCCN和ESMO指南的推荐意见。
从更宏观的视角审视,Amivantamab皮下制剂的成功开发标志着生物制剂竞争维度从单纯的疗效和安全性向”给药便利性+患者体验+医疗资源效率”多元化拓展。对于MARIPOSA方案(Amivantamab+Lazertinib)而言,SC制剂的批准显著改善了其相对口服TKI的便利性短板——月度皮下注射(7分钟给药时间)与每日口服之间的体验差距已大幅缩小,这可能影响一线治疗中MARIPOSA方案与奥希替尼单药(或FLAURA-2方案)的竞争格局。未来,皮下制剂技术的推广有望扩展到更多生物制剂(如其他双特异性抗体和ADC),成为肿瘤精准治疗领域制剂创新的重要方向。