


ICH E3 长解读版 | 临床开发视角
基于 ICH E3 原文结构和 20 张信息图,从临床开发实战角度拆解 CSR 的写作逻辑、审评关注点和证据链要求。
ICH E3《Structure and Content of Clinical Study Reports》规定的是单个临床研究报告的结构和内容。它不是单纯的医学写作指南,也不是把统计结果整理成论文,而是把一项临床研究从科学问题、伦理合规、方案设计、实施过程、受试者流向、疗效和安全性结果,到表图、listing、CRF 和附录归档,组织成监管可以审评、可以复核、可以追溯的完整证据包。站在临床开发视角,E3 的价值在于帮助团队从研究一开始就倒推最终 CSR 所需证据:哪些设计要解释,哪些执行偏差要记录,哪些医学判断要透明,哪些数据必须能追溯到源记录。后台发送ICHE3 获取官方中文 pdf 和图片幻灯
01. 一图总览:CSR 是监管可接受的临床研究完整证据包
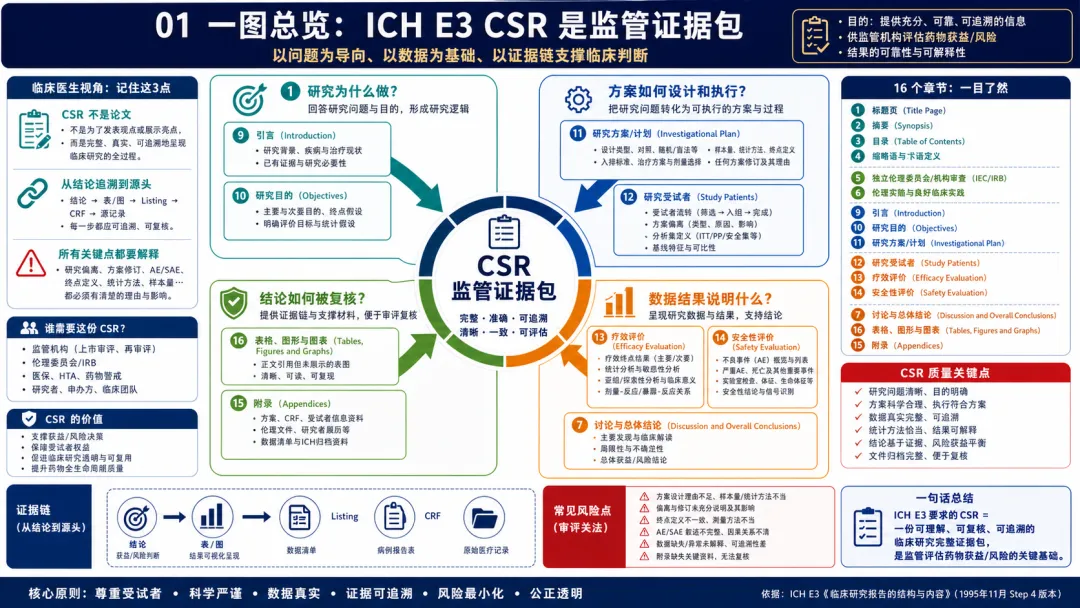
图 01 01. 一图总览:CSR 是监管可接受的临床研究完整证据包
解读:ICH E3 首先要我们改变对 CSR 的理解:它不是研究结束后写出来的一篇“总结文章”,而是一项临床研究面向监管审评的完整证据包。原文把 CSR 拆成标题页、摘要、伦理、研究者结构、研究目的、研究方案、受试者、疗效、安全性、讨论、表图、参考文献和附录等部分,本质上是在建立一条从研究问题到监管判断的证据链。站在临床开发视角,CSR 必须同时回答四类问题:为什么开展这项研究,研究如何设计并实际执行,数据说明了什么,结论是否能被表图、listing、CRF 和源记录复核。很多团队把 CSR 当成医学写作部门的任务,这是错误的。CSR 质量往往由研究执行阶段决定:入排标准是否记录清楚,终点评估是否一致,AE/SAE 叙述是否完整,方案偏离和修订是否说明原因与影响。E3 的核心精神是透明、完整和可追溯。对临床开发人员来说,读 E3 不是为了背章节,而是为了在方案设计、数据管理、医学监查和数据库锁库前就知道最终报告会怎样审视这项研究。 进一步看,这张总览图也适合作为项目启动时的培训页:让医学、运营、数据管理、统计和写作团队先形成同一张“终局地图”,明确每个部门交付的文件和数据最终会落到 CSR 哪个位置,避免研究结束后才发现证据链断裂。
02. 标题页、摘要、目录与术语:让审评者快速定位研究

图 02 02. 标题页、摘要、目录与术语:让审评者快速定位研究
解读:E3 前四章看似偏行政和格式,但在监管审评中非常关键。标题页要交代研究标题、方案编号、版本日期、申办方、研究者和签名等信息,它解决的是“这份报告对应哪项研究、由谁负责、版本是否明确”的问题。摘要则是审评者快速把握研究全貌的入口,原文要求摘要覆盖研究目的、方法、受试者、治疗、疗效和安全性结果以及结论。临床开发人员要特别注意,摘要不是选择性展示亮点,也不是市场化表达,而是高度压缩但不能失真的研究快照。目录和术语定义同样重要。目录让审评者能快速定位关键证据,缩略语和术语则保证全篇口径一致。很多 CSR 的问题不是数据本身,而是同一概念在方案、SAP、表格和正文中叫法不一致,例如分析集、终点、访视窗口、AE 口径或停药原因。E3 通过这些“入口导航”章节提醒我们:一份可审评的 CSR,首先必须让读者知道它是谁、讲什么、结构在哪里、术语如何理解。对临床开发团队而言,这些内容应在报告写作前就与方案、SAP、数据集和 TLF 标准保持一致。 因此,临床开发团队在启动 CSR 前,应先统一标题页信息、方案版本、SAP 版本、数据库锁定日期和主要表图目录。摘要尤其需要医学、统计和安全团队共同审阅,确保它既能快速呈现研究全貌,又不跳过关键限制和重要安全发现。
03. 伦理、知情同意与研究者结构:CSR 的合规地基
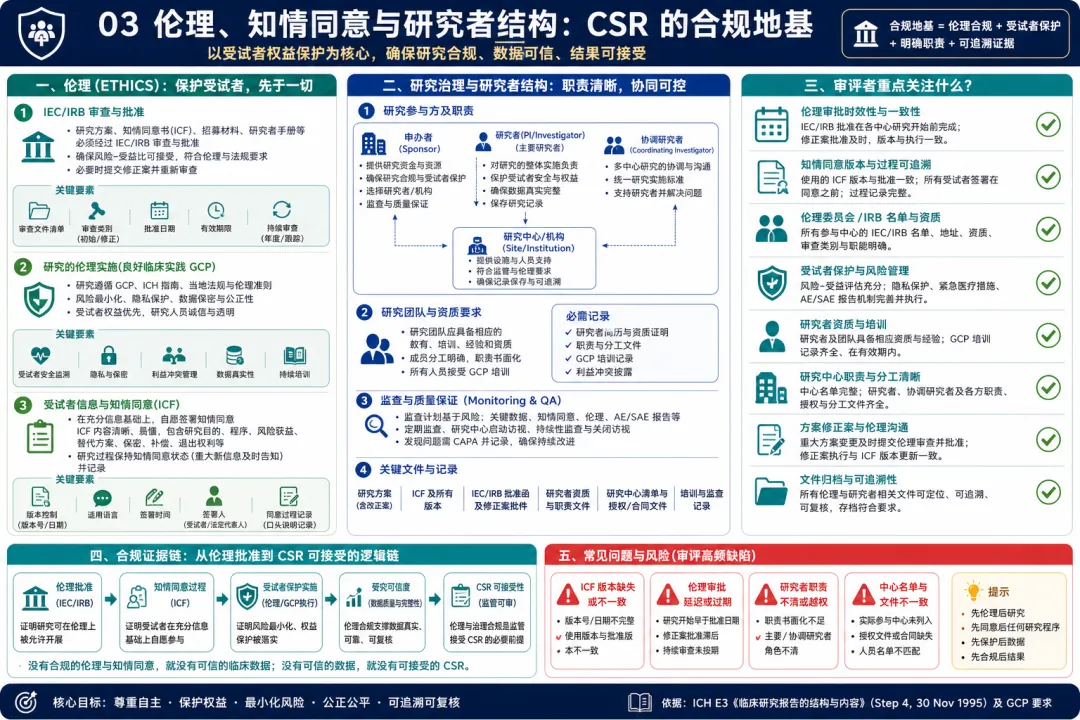
图 03 03. 伦理、知情同意与研究者结构:CSR 的合规地基
解读:E3 第5章和第6章把伦理和研究治理放在结果之前,这体现了监管审评的基本逻辑:临床研究结果再有吸引力,也必须建立在受试者保护和合规执行的基础上。原文要求说明独立伦理委员会或机构审查委员会、研究的伦理实施、患者信息和知情同意,以及研究者和研究行政结构。站在临床开发视角,这些内容不是“后台文件清单”,而是证明研究可信度的重要部分。比如,伦理批准日期是否覆盖研究启动时间,知情同意版本是否与方案修订一致,受试者是否在任何研究程序前完成知情同意,中心和研究者职责是否清楚,协调研究者或主要研究者签名是否完整。对于多中心国际研究,研究治理结构尤其重要:谁负责医学判断,谁负责中心执行,谁负责数据质量,谁对最终报告负责,都需要清楚呈现。实践中常见问题包括知情同意版本混乱、伦理批准材料不完整、研究者名单与实际入组中心不一致、关键职责没有文件化。E3 提醒临床开发团队,CSR 不只是疗效和安全性报告,也是在向监管证明:这项研究是在受试者权益受到保护、职责边界清楚、伦理流程可核查的条件下完成的。 从项目管理角度,伦理和研究者结构还应与 TMF、中心启动包、授权分工表和监查记录保持一致。CSR 写作阶段如果发现这些材料互相矛盾,往往说明研究执行证据本身存在缺口,需要先回到文件系统和中心记录中完成核对。
04. 引言与研究目的:把临床问题写成可审评问题
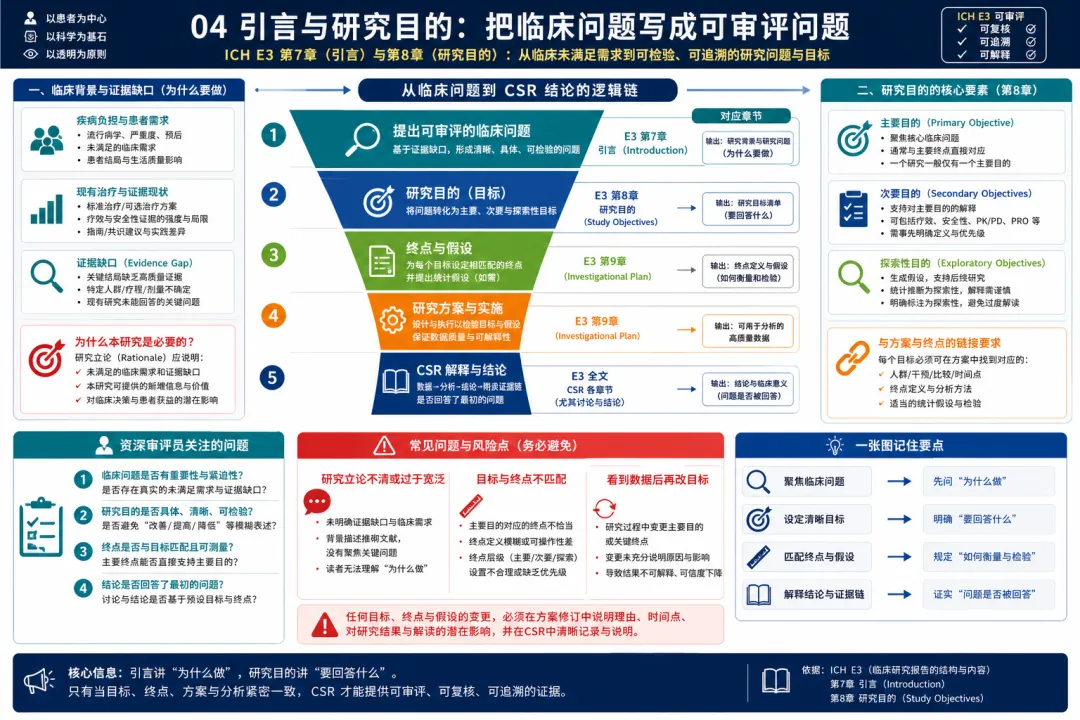
图 04 04. 引言与研究目的:把临床问题写成可审评问题
解读:E3 第7章和第8章要求报告引言和研究目的。引言要把研究放进疾病背景、现有治疗、药物开发阶段和证据缺口中;研究目的则要明确这项研究具体想回答什么问题。临床开发中,一个常见误区是把“研究目的”写成宽泛口号,例如评价疗效和安全性,但没有说明主要问题、目标人群、比较方式和预期临床价值。E3 的逻辑是:研究目的必须能向后连接到设计、终点、样本量和统计方法,也必须能向前回应未满足需求或开发策略。对于早期研究,目的可能是探索剂量、安全性、PK/PD 或初步疗效;对于确证性研究,目的则需要支撑注册决策和获益风险判断。CSR 中的引言和目的不是装饰性背景,而是后续结果解释的参照系。如果研究目的不清楚,主要终点为何选择、对照组为何合理、分析集为何定义、结论为何成立都会变得模糊。站在临床开发视角,写 CSR 前应回到原始方案和开发计划,确认研究目的是否在研究执行过程中发生变化,任何变化是否有方案修订和统计计划支持。好的 E3 式写法,是让审评者在进入结果前就理解:这个研究问题有临床意义,设计选择有理由,结论将围绕预设问题展开。 对新手临床开发人员而言,这一部分还提示一个实用原则:不要先写结果再倒推目的。真正稳健的写法是从未满足需求和开发策略出发,说明研究目的,再解释设计和终点,最后让结果自然回答这些预设问题。
05. Investigational Plan 总览:第9章是 CSR 的骨架
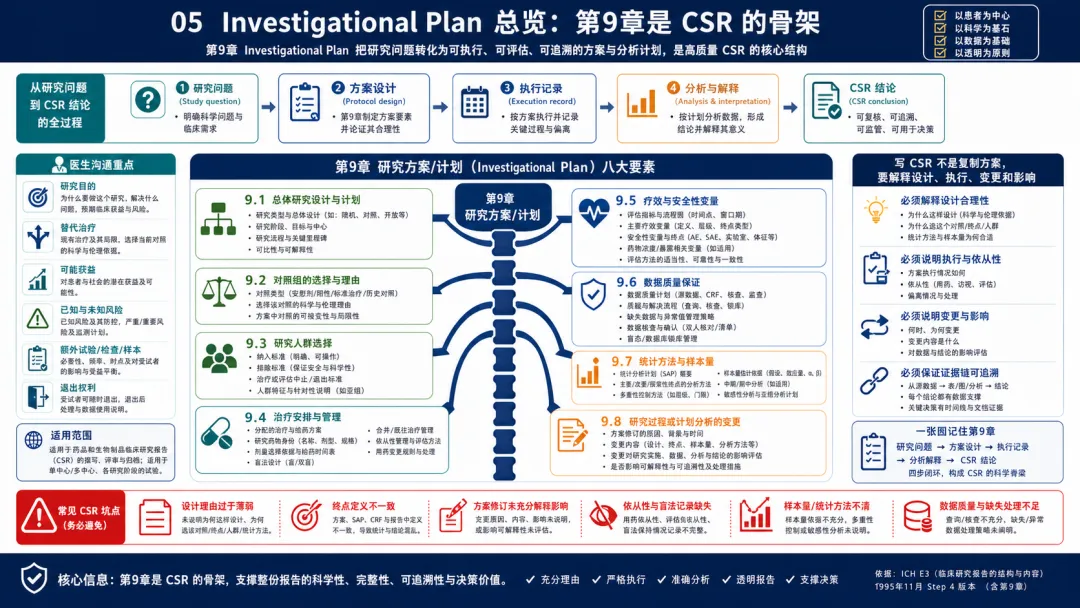
图 05 05. Investigational Plan 总览:第9章是 CSR 的骨架
解读:E3 第9章是整份 CSR 的骨架。原文要求报告总体研究设计、对照组选择、人群选择、治疗方案、疗效和安全性变量、数据质量保证、统计方法和样本量,以及研究实施或计划分析的任何变化。临床开发人员应把第9章理解为“方案如何转化为可审评证据”的章节,而不是把方案复制粘贴进 CSR。这里真正需要说明的是:为什么采用这种设计,为什么选择这种对照,为什么纳入这类患者,为什么这样给药和评估,哪些变量用于回答主要问题,数据质量如何保证,统计分析如何预先定义,以及实际执行中发生了哪些影响解释的变化。审评者会用第9章作为参照,去判断后面的患者流向、疗效结果和安全性结论是否可信。实践中很多 CSR 的弱点也集中在第9章:设计理由过薄,方案修订没有说明影响,终点定义和 SAP 不一致,盲法或依从性记录缺失,样本量假设讲不清楚。站在临床开发视角,第9章要求团队在方案设计、医学监查、数据管理和统计之间形成一致叙事。CSR 不是只呈现“发生了什么”,还要解释“为什么这样设计、实际是否这样执行、偏离或变更是否改变了结论解释”。 因此,项目进入 CSR 阶段前,最好把第9章作为跨职能核对清单使用:医学确认设计和治疗逻辑,运营确认执行过程,数据管理确认质量流程,统计确认分析计划,安全团队确认变量和事件收集口径。
06. 总体设计与对照组:为什么这样设计、为什么这样比较

图 06 06. 总体设计与对照组:为什么这样设计、为什么这样比较
解读:E3 9.1 和 9.2 要求说明总体研究设计和对照组选择,包括研究类型、设计结构、研究流程、中心、周期、随机化、盲法以及对照组的科学和伦理理由。临床开发视角下,这一部分直接关系到疗效和安全性结论的可解释性。不同设计解决不同问题:安慰剂对照有助于检测药物效应,但必须满足伦理可接受性;阳性对照可以贴近临床实践,但需要考虑 assay sensitivity 和非劣效界值;剂量对照适合剂量反应探索;开放标签设计则要特别处理偏倚风险。E3 要求讨论研究设计,包括对照组选择,实际上是在要求申办方解释“为什么这个比较能回答研究目的”。如果对照组不恰当,后续即便出现统计学差异,也可能难以转化为明确临床判断。对临床开发团队来说,CSR 中应清楚写出设计选择如何对应研究阶段和开发目标,访视和评估窗口如何减少偏倚,随机化和盲法如何保护客观性,哪些限制可能影响外推。常见风险包括对照组选择理由过于简短、开放标签对主观终点影响未讨论、研究流程图与实际执行不一致、治疗转换或救援治疗未解释。E3 的提醒很直接:设计不是背景信息,而是结果可信度的前提。 在撰写时,还应把设计限制说清楚,而不是回避。例如开放标签、交叉、救援治疗、入组慢、中心差异或对照治疗变化,都可能影响解释。主动说明限制和缓解措施,通常比在审评问询中被动解释更有利。
07. 研究人群与治疗安排:入排、退出、给药、盲法和依从性

图 07 07. 研究人群与治疗安排:入排、退出、给药、盲法和依从性
解读:E3 9.3 和 9.4 聚焦研究人群和治疗安排。人群部分包括入选标准、排除标准,以及受试者从治疗或评估中移除的规则;治疗部分包括治疗分配、研究药物身份、剂量选择、每位受试者给药时间、盲法、既往和合并治疗、治疗依从性。站在临床开发视角,这些信息决定两个核心问题:研究结果适用于哪些患者,以及受试者实际接受的治疗是否支持结果解释。入排标准过窄会限制外推,过宽可能增加异质性;退出规则不清会影响终点可评价性;剂量和给药时点记录不完整会影响暴露-反应和安全性分析;合并用药和依从性处理不清会使疗效信号难以解释。E3 要求在 CSR 中详细说明这些内容,是为了让审评者判断研究执行是否与方案一致,实际治疗暴露是否足以支持结论。临床医生在这里的作用非常关键:入排判断、合并疾病、既往治疗、停药原因、剂量调整、漏服、盲法破盲、合并用药的医学理由,最终都会进入 CSR 的解释链。实践中常见缺陷包括退出原因写得过于笼统,依从性只报百分比不解释异常,剂量调整缺少日期和原因,禁止用药未被及时记录。E3 让我们看到,现场医学记录质量直接决定 CSR 的可信度。 对医生来说,这张图也提醒现场记录要有“未来可解释性”。一次停药、一次剂量调整、一次退出评估,如果只写“医生决定”或“患者原因”,在 CSR 中几乎没有解释价值;应尽量记录具体医学原因、时间点和后续处理。
08. 疗效安全变量与数据质量:测什么、怎么测、数据是否可靠

图 08 08. 疗效安全变量与数据质量:测什么、怎么测、数据是否可靠
解读:E3 9.5 和 9.6 讨论疗效和安全性变量以及数据质量保证。原文要求说明被评估的疗效和安全性测量、评估流程图、测量方法适当性、主要疗效变量、药物浓度测量,以及数据质量保证措施。临床开发中,这一部分经常被低估,但它决定了研究能否从“收集数据”变成“产生可信证据”。首先,变量必须与研究目的匹配:主要终点要能回答核心问题,安全变量要覆盖预期和未知风险,评估时间点要能捕捉药物作用和风险窗口。其次,测量方法必须适当:影像评估是否有中心阅片,量表是否经过培训,实验室是否标准化,PK 样本时间是否准确,安全访视是否覆盖高风险期。第三,数据质量保证必须能说明监查、数据核查、疑问管理、质量控制和数据库锁定前的关键流程。对临床开发团队来说,E3 的要求意味着终点定义、CRF 设计、数据管理计划、医学监查计划和 SAP 不能割裂。常见问题包括终点定义前后不一致,访视窗口执行松散,缺失数据原因不清,实验室单位或参考范围混乱,影像或量表评价没有质量控制。CSR 中如果不能说明数据如何产生和如何保证质量,后续再复杂的统计分析也难以弥补。 这也是为什么终点评估培训和数据清理不能只靠数据管理完成。医学团队需要持续关注关键变量的缺失、异常和口径漂移,及时判断这些问题是否会影响主要终点、安全信号或受试者层面的医学解释。
09. 统计方法、样本量与研究变化:计划如何影响结论解释
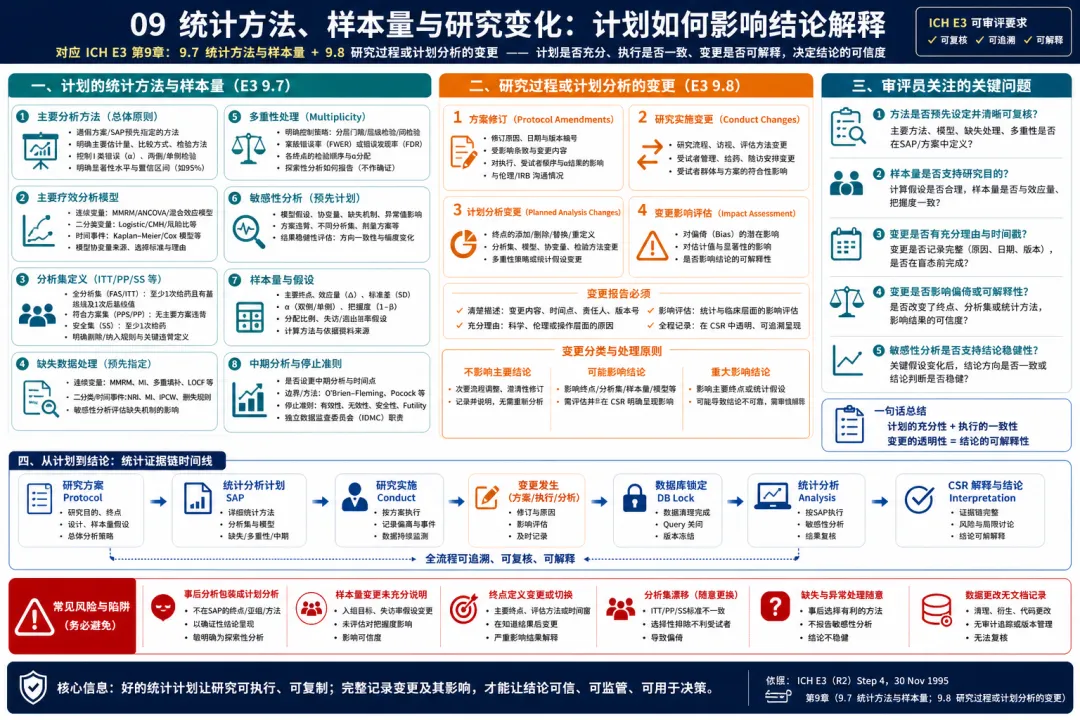
图 09 09. 统计方法、样本量与研究变化:计划如何影响结论解释
解读:E3 9.7 和 9.8 要求报告方案中计划的统计方法、分析计划、样本量确定依据,以及研究实施或计划分析中的任何变化。临床开发视角下,这一部分是 CSR 透明度的关键测试。统计方法不是报告末尾的技术细节,而是研究从数据到结论的推理规则。分析集如何定义,主要分析模型是什么,缺失数据如何处理,多重性如何控制,协变量是否预设,亚组和敏感性分析如何安排,样本量假设来自哪里,这些都直接决定结论是否可信。E3 还特别要求说明研究实施或计划分析的变化,这一点非常重要。真实研究中常会出现方案修订、样本量调整、终点定义细化、访视窗口改变、入组策略调整或 SAP 更新。问题不在于是否发生变化,而在于变化是否有时间线、理由、批准路径以及对结果解释的影响评估。监管审评最敏感的是“看完数据后再改规则”的风险。因此,CSR 必须把预设计划和实际执行区别清楚。临床开发团队在锁库前应系统梳理所有方案修订、SAP 版本、关键决策会议纪要和偏离分类,确保报告中对变更的叙述完整、诚实、可追溯。 实战中建议建立“变更索引表”,把方案修订、SAP 更新、重要运营决策、数据处理规则变化和盲态/非盲态讨论分开记录。CSR 写作时再逐项判断哪些需要进入正文,哪些放入附录,哪些需要在讨论中解释。
10. 研究受试者:患者流向和方案偏离是结果解释的入口

图 10 10. 研究受试者:患者流向和方案偏离是结果解释的入口
解读:E3 第10章要求报告研究受试者,包括受试者处置和方案偏离。它是结果章节的入口,因为在看疗效和安全性之前,审评者必须先知道有哪些患者进入研究、接受治疗、完成评估、提前退出或被排除出分析。受试者流向不仅是 CONSORT 图式的展示,更是对研究执行完整性的检验。筛选失败、随机化、治疗暴露、完成访视、停药、中止、退出随访,每一步都应有清楚分母、原因和时间点。方案偏离同样重要。E3 要求报告偏离,是因为偏离可能影响受试者权益、数据完整性、分析集归属和终点可评价性。临床开发人员应避免把偏离当成质量部门的孤立问题。一个关键入排偏离可能改变人群解释,一个终点评估窗口偏离可能影响疗效评价,一个禁止合并用药偏离可能影响安全性或疗效归因。CSR 中应清楚说明偏离类型、严重程度、发生原因、涉及受试者数量、对分析和解释的影响。常见问题包括退出原因分类过粗,偏离列表与分析集排除不一致,重要偏离没有医学解释,患者流向图和正文数字不一致。E3 提醒我们:患者流向和偏离管理,是审评者理解结果可信度的第一道门。 因此,研究进行中就应维护患者流向和重要偏离的动态清单,而不是等锁库后再整理。医学团队应提前识别可能影响主要分析或安全解释的偏离,并与统计团队确认其对分析集和敏感性分析的影响。
11. 疗效评价基础:分析集、基线特征和依从性

图 11 11. 疗效评价基础:分析集、基线特征和依从性
解读:E3 11.1 到 11.3 规定疗效评价的基础内容:分析的数据集、人口学和其他基线特征、治疗依从性。临床开发视角下,疗效结果不是从主要终点表开始的,而是从“谁被分析、他们是否可比、是否实际接受了足够治疗”开始。分析集定义决定结论适用于哪些受试者。ITT 或 FAS 更强调随机化后的整体效应,PPS 更强调符合方案人群,安全集则通常与实际用药有关。CSR 必须说明每个分析集的定义、人数、排除原因和与方案/SAP 的一致性。基线特征则帮助判断组间可比性和外推价值,包括年龄、性别、疾病严重程度、诊断依据、既往治疗、预后因素和关键生物标志物。依从性看似操作性指标,但会影响疗效暴露和结果解释,尤其在口服药、长期治疗或剂量调整频繁的研究中更关键。对临床医生来说,这些内容来自日常诊断、病史采集、用药记录和访视管理。常见风险包括分析集口径漂移,基线不平衡未讨论,排除受试者原因不透明,依从性异常但未解释对疗效的影响。E3 的要求让我们看到,疗效评价的可信度建立在分析集、基线和实际治疗暴露的共同基础上。 对项目团队来说,这一章也是发现数据问题的早期窗口。如果分析集人数、基线表、依从性表和患者流向图之间不一致,通常不是排版问题,而是数据口径、排除规则或程序执行需要重新核对。
12. 疗效结果:主要终点、次要终点、亚组和敏感性

图 12 12. 疗效结果:主要终点、次要终点、亚组和敏感性
解读:E3 11.4 是疗效结果的核心章节,要求展示疗效结果、统计和分析问题、协变量调整、退出和缺失处理、多重性、疗效子集、等效或非劣效研究、亚组检查、个体反应数据、剂量或浓度与反应关系、相互作用、按患者展示以及疗效结论。临床开发人员应特别注意,E3 并不鼓励只报告一个显著 P 值。CSR 要做的是把疗效发现放进预设分析框架中解释:主要终点是否达到,次要终点是否支持,敏感性分析是否一致,亚组发现是否可信,缺失数据是否可能改变结论,个体数据是否存在异常模式。对于注册性研究,还要区分统计显著性和临床意义;对于非劣效或等效研究,要清楚说明界值、分析集和敏感性;对于剂量探索或早期研究,则要关注剂量-反应、浓度-反应和疗效持续性。临床医生在疗效章节中的贡献不只是确认数据,还包括解释异常反应、疾病进展、评估偏差、合并治疗和终点临床意义。常见问题包括只展示有利亚组,忽略多重性,缺失数据处理与 SAP 不一致,个体患者数据无法支持汇总结论。E3 的精神是:疗效结论必须由预设方法、汇总结果和个体证据共同支撑。 在临床开发决策中,疗效章节还要服务于下一步开发:是否支持剂量选择,是否支持目标人群收窄,是否需要补充研究,是否足以支撑注册沟通。CSR 写法应让这些判断有证据基础,而不是只给出统计结论。
13. 安全性总览:暴露、不良事件和安全风险识别
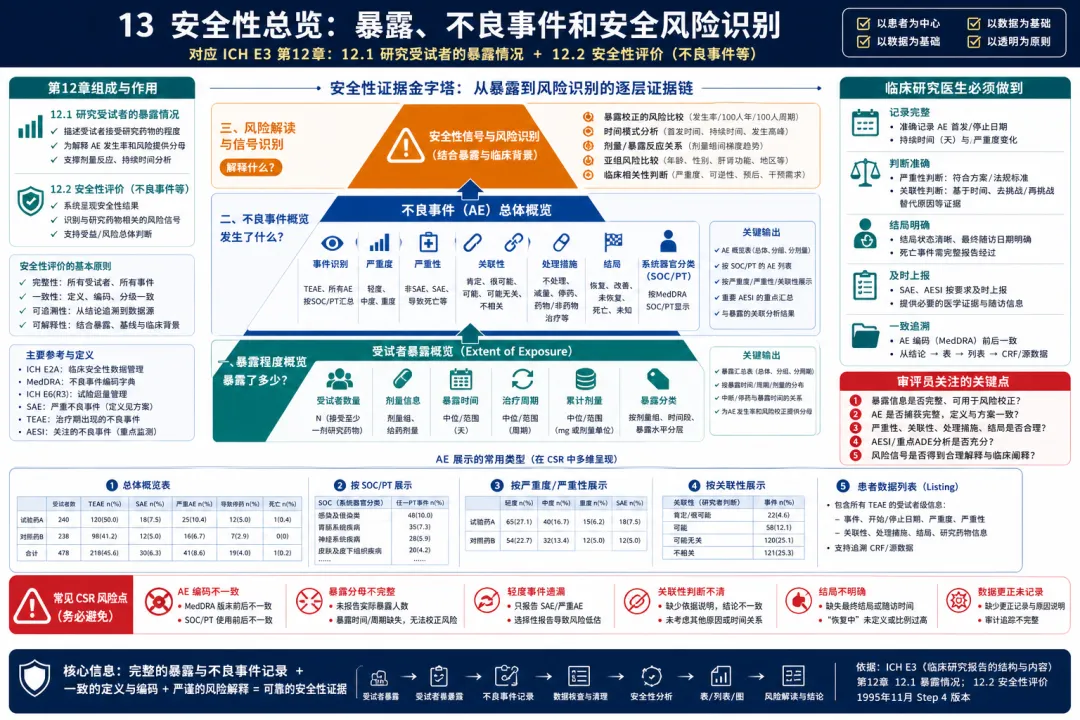
图 13 13. 安全性总览:暴露、不良事件和安全风险识别
解读:E3 第12章从安全性评价开始时,首先要求报告暴露程度,再报告不良事件。这一顺序非常有意义:没有暴露背景,就无法解释安全性风险。临床开发人员在 CSR 中不仅要说明有多少受试者接受治疗,还要说明剂量、治疗持续时间、周期数、累计暴露、剂量调整和停药情况。随后,AE 的展示要覆盖总体概况、按事件类别和患者列表展示、严重程度、严重性、与研究药物的关系、处理措施和结局。安全性分析不是简单比较 AE 发生率,而是结合暴露、时间关系、剂量关系、风险人群、事件严重程度和临床意义来识别风险信号。对医生而言,AE 记录质量是安全章节的生命线。起止日期、严重程度、严重性标准、因果关系、处理措施、转归、是否导致停药或剂量调整,都必须完整。实践中常见问题包括 AE 编码前后不一致,暴露分母不清,轻中度事件记录不足,因果关系判断缺少医学理由,AE 与实验室异常或 SAE 叙述之间不一致。E3 的要求帮助团队把安全性从“事件列表”提升为系统风险识别:药物在什么暴露下、对哪些患者、以什么模式产生了哪些风险。 对安全团队而言,暴露数据和 AE 数据应同步审阅。只有知道患者实际接受了多少治疗、治疗多久、是否减量或停药,才能判断某类 AE 的发生率、严重性和临床意义是否构成真正风险信号。
14. 死亡、SAE 和显著不良事件:叙述性病例是安全判断核心

图 14 14. 死亡、SAE 和显著不良事件:叙述性病例是安全判断核心
解读:E3 12.3 对死亡、其他严重不良事件和其他显著不良事件提出专门要求,包括列表、叙述和分析讨论。这些事件往往是监管判断获益风险的核心。与普通 AE 汇总不同,死亡和 SAE 需要通过个案叙述呈现完整医学故事:患者背景、疾病状态、既往病史、研究药物暴露、事件发生时间线、相关检查、处理过程、停药或减量、结局、研究者因果判断和申办方医学评估。临床开发人员要理解,叙述性病例不是简单把数据库字段拼接成段落,而是要帮助审评者判断事件是否可能与药物相关,是否存在共同模式,是否提示风险管理需求。尤其在肿瘤、心血管、感染或重症研究中,死亡和 SAE 可能与基础疾病、进展、合并治疗和研究药物多因素相关,更需要清楚医学推理。E3 还要求对这些事件进行分析和讨论,这意味着不能只提交个案叙述,还要在总体层面识别是否有聚集趋势、器官系统模式、剂量关系或时间关系。常见问题包括时间线缺失,事件结局不完整,因果判断没有理由,叙述与 AE 表格不一致,病例间模式没有讨论。高质量 CSR 的安全性判断,往往取决于这些关键病例叙述是否扎实。 建议在 CSR 写作前召开关键病例医学复核会,逐例确认死亡、SAE、AESI 和导致停药事件的叙述是否完整,是否与数据库、叙述稿、表格和药物警戒系统一致,避免提交后出现难以解释的矛盾。
15. 实验室、生命体征和安全结论:从异常值到临床意义

图 15 15. 实验室、生命体征和安全结论:从异常值到临床意义
解读:E3 12.4 到 12.6 要求报告实验室检查、生命体征、体格检查和其他安全观察,并形成安全性结论。临床开发视角下,实验室和体征数据的难点不是数量,而是临床意义判断。CSR 需要区分随机波动、疾病相关变化、合并用药影响和潜在药物风险。实验室部分通常需要汇总变化趋势、异常值、按严重程度或等级转移、个体临床显著异常,以及必要的患者列表。生命体征、体格检查、ECG 或其他专项安全评估也应遵循类似逻辑:先看总体趋势,再看异常个体,最后判断是否具有临床意义和药物相关性。医生在这一部分的判断非常关键。例如,一个 ALT 升高是否需要结合基线肝病、合并用药、胆红素、停药后恢复和再暴露情况;一个 QT 延长是否需要结合电解质、心率、合并药物和中心阅片;一个血压变化是否与剂量或时间相关。E3 要求最终形成安全性结论,意味着不能只堆表格,而要说明是否出现安全信号、信号强度如何、哪些证据支持或削弱因果关系、哪些不确定性仍存在。常见缺陷包括异常值列表完整但没有医学解释,缺少基线比较,缺少复测或转归,安全结论与表格发现脱节。 对实验室和体征数据,团队还应预先定义哪些异常需要个案叙述或医学解释,哪些只需汇总展示。这样可以避免报告阶段临时判断标准不一致,也能让安全结论更清楚地区分噪音和真实风险。
16. 讨论与总体结论:把数据转化为临床判断
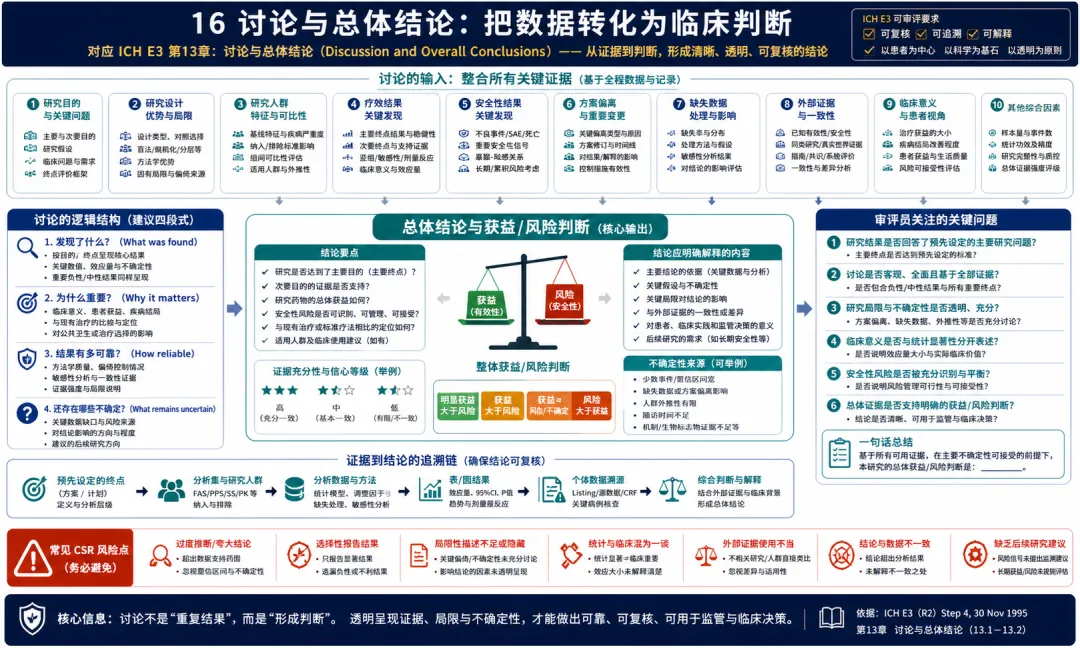
图 16 16. 讨论与总体结论:把数据转化为临床判断
解读:E3 第13章要求讨论和总体结论。站在临床开发视角,这是 CSR 从数据报告走向医学判断的关键章节。讨论不是重复疗效和安全性结果,而是把研究目的、设计、执行质量、患者人群、疗效一致性、安全风险、方案偏离、缺失数据和研究局限放在一起解释。一个成熟的讨论章节应回答:研究是否达成预设目的,主要终点和次要终点是否形成一致证据,疗效大小是否具有临床意义,安全性风险是否可接受,哪些发现需要谨慎解释,哪些不确定性可能影响外推。E3 的结构安排也提示我们,讨论必须建立在前面章节完整证据之上。没有清楚的方案设计,就无法评价结果可靠性;没有患者流向和偏离分析,就无法判断偏倚;没有安全性叙述和表图,就无法做获益风险判断。临床开发团队在写讨论时应避免两类极端:一是把讨论写成宣传材料,只强调积极结果;二是机械重复统计输出,没有临床解释。监管审评需要的是平衡、透明、可追溯的判断。常见问题包括过度解读亚组,忽略阴性或不一致结果,对缺失数据和方案偏离轻描淡写,安全风险与疗效收益没有放在同一框架下讨论。E3 的总体结论应服务于科学决策,而不是替代证据本身。 讨论章节最好由医学负责人牵头,但不能只由医学写作单独完成。它需要统计、安全、数据管理和临床运营共同确认事实基础。任何超出数据支持范围的表达,都应被删减或改写为更审慎的判断。
17. 表、图和图形:正文之外的证据展示层

图 17 17. 表、图和图形:正文之外的证据展示层
解读:E3 第14章规定正文引用但未纳入正文的表、图和图形,包括人口学、疗效和安全性数据。临床开发人员应把表图理解为 CSR 证据链的展示层,而不是报告排版附件。正文提出的每一个关键判断,都应能在表图中找到清楚支撑;表图中的每一个汇总数字,又应能继续追溯到 listing 和原始记录。高质量表图有几个共同特征:标题清楚,分析集明确,分母一致,访视或时间点明确,单位和统计方法标注完整,缺失数据和异常处理有说明,脚注能解释关键规则。对于疗效数据,表图要支持主要终点、次要终点、敏感性分析、亚组和个体反应的解释;对于安全性数据,表图要覆盖暴露、AE、SAE、死亡、显著事件、实验室异常和生命体征。实践中,很多 CSR 问题来自正文、表格、listing 三者不一致。例如正文说某事件罕见,但 AE 表显示聚集趋势;正文引用某分析集,但表格使用不同分母;图形展示的访视窗口与 SAP 不一致。E3 通过第14章提醒团队,表图不是越多越好,而是要让审评者能快速、准确、可复核地理解证据。TLF 开发应尽早与 CSR 叙事、SAP 和数据标准同步。 因此,TLF 审阅不应只检查格式,还应检查叙事一致性。每张关键表图都要问三个问题:它支持正文哪句话,分母和分析集是否正确,是否能继续追溯到 listing 和患者级数据。
18. 附录16.1:方案、CRF、伦理和研究者资料

图 18 18. 附录16.1:方案、CRF、伦理和研究者资料
解读:E3 16.1 是研究信息附录,内容包括方案和方案修订、样例 CRF、伦理委员会和知情同意材料、研究者和关键参与者信息、签名、随机化方案和编码、审计证书、统计方法文件、实验室标准化和质量保证文件、基于研究的发表以及重要参考文献等。临床开发中,附录常被视为“最后整理的文件夹”,但在监管审评中它是复核研究真实性和执行一致性的底层证据。方案和修订让审评者看到规则如何演变;CRF 显示数据如何被收集;伦理和知情同意材料证明受试者保护;研究者资质和签名体现责任归属;随机化和盲法文件支持偏倚控制;实验室标准化文件支持数据可比性。对于临床开发团队,16.1 的挑战在于版本控制和影响解释。每一次方案修订是否有日期、理由、伦理批准和实施范围?CRF 是否与实际数据收集一致?知情同意是否对应正确版本?研究者和中心名单是否完整?这些问题如果在研究结束后才回头补,会非常被动。E3 的附录要求提醒我们,CSR 不是单一 Word 文档,而是一个证据包。项目管理、临床运营、医学、统计、数据管理和质量部门都应在研究过程中维护这些证据,确保最终报告能够被审评者逐层复核。 从运营角度,16.1 附录最好在研究执行期间持续维护,而不是 CSR 写作时临时收集。版本、日期、签名、批准路径和适用中心一旦缺失,后期补救成本很高,也容易引发审评中的一致性问题。
19. 附录16.2-16.4:患者数据列表、CRF 和归档 listing

图 19 19. 附录16.2-16.4:患者数据列表、CRF 和归档 listing
解读:E3 16.2 到 16.4 关注患者数据 listing、关键 CRF 和个体患者数据归档。原文列出的 listing 包括中止患者、方案偏离、被排除出疗效分析的患者、人口学数据、依从性或药物浓度、个体疗效反应、AE 列表、必要时的个体实验室测量等;CRF 则特别强调死亡、SAE 和因 AE 退出等关键病例。临床开发视角下,这部分是 CSR 可追溯性的底层。正文提供叙述,表图提供汇总,listing 提供患者级数据,CRF 和源记录提供原始证据。如果某个主要终点结果无法追到个体反应列表,某个 SAE 叙述无法追到 CRF,某个实验室异常无法追到原始测量,那么 CSR 的可信度就会下降。这里也体现了医生记录的重要性。医学判断、事件时间线、停药原因、疾病进展、合并用药、终点评估依据,如果只在现场口头讨论而没有进入可追溯记录,最终很难在 CSR 中被审评者接受。常见问题包括 listing 缺少关键变量,患者编号或访视日期不一致,CRF 与数据库字段不匹配,隐私处理和可追溯性之间没有平衡,归档 listing 与提交表格不一致。E3 要求团队建立从患者到结论的完整链条:源记录支持 CRF,CRF 支持数据集,数据集支持表图,表图支持正文结论。 对临床开发团队来说,listing 审阅不是数据管理的单独任务。医学团队需要抽查关键患者的 listing 与病例叙述、CRF 和源记录是否一致,尤其是影响主要终点、安全事件、退出和方案偏离的病例。
20. 从 E3 到实战:医生如何为高质量 CSR 做准备
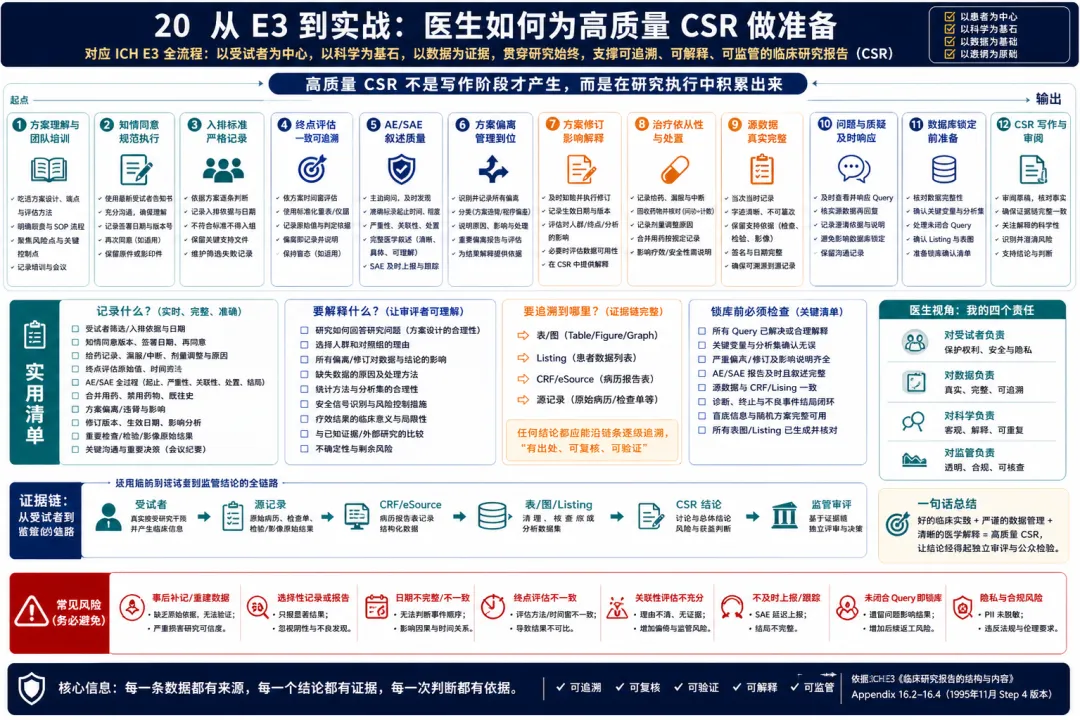
图 20 20. 从 E3 到实战:医生如何为高质量 CSR 做准备
解读:把 E3 用到临床开发实战中,最重要的转变是:不要等研究结束后才考虑 CSR,而要在方案设计和研究执行阶段就按最终证据链倒推。医生和临床开发团队需要提前想清楚几个问题。第一,研究目的、终点、人群和对照组是否能形成一致逻辑;第二,知情同意、伦理批准、入排判断、治疗暴露、终点评估、AE/SAE 叙述、方案偏离和修订是否都能被记录和追溯;第三,数据库、表图、listing 和 CRF 是否能支持正文要写出的临床结论;第四,安全性和疗效的医学解释是否透明,是否能说明不确定性和局限。高质量 CSR 的准备工作贯穿全程:方案阶段要保证设计可解释,启动阶段要培训关键记录要求,执行阶段要及时处理偏离和医学疑问,数据清理阶段要关注影响结论的关键变量,锁库前要确认分析集、终点、AE、SAE、实验室异常和重要病例叙述,报告阶段要把证据组织成一致叙事。对临床试验医生来说,E3 是一份非常实用的“终局视角”文件:它告诉我们,今天在病历、CRF、医学判断和沟通记录中留下的内容,都会决定未来 CSR 是否可信、审评是否顺畅、获益风险判断是否站得住。 因此,E3 可以被用作研究全生命周期的质量工具。项目团队如果在方案设计、启动培训、监查、数据清理、锁库和 CSR 审阅各阶段都按 E3 倒推证据需求,最终报告会更顺畅,审评问询也会更少。
总结
从临床开发视角看,ICH E3 的真正价值在于把 CSR 从“写作产物”提升为“研究证据系统”。一项研究最终能否被清楚审评,不只取决于疗效结果是否阳性,也取决于研究目的是否明确、方案是否合理、执行是否一致、偏离是否透明、数据是否可靠、安全性判断是否充分、附录证据是否完整。对资深临床试验者来说,E3 是研究全生命周期的倒推工具:用最终 CSR 的结构反推方案设计、现场记录、医学监查、数据管理和统计分析。高质量 CSR 不是最后写出来的,而是在研究设计和执行过程中被一点点积累出来的。
资料来源:ICH E3 Structure and Content of Clinical Study Reports, Step 4 version dated 30 November 1995