



|中肽生化内容团队编辑
过去数年,大环肽(macrocyclic peptides)正在成为新药研发中最受关注的分子类别之一。它们不同于传统线性多肽——通过环化赋予更刚性的构象、更高的靶点选择性与更强的代谢稳定性,部分分子甚至突破了"多肽不能口服"的经典认知,在心血管、免疫、肿瘤等领域显示出真实的临床价值。
然而,与生物学潜力相伴随的,是极高的合成门槛。环肽往往结构高度复杂:多个手性中心、非天然氨基酸、多步关环反应、严苛的立体化学控制,加之工业化生产对收率和纯化方式的强制约,使得这类分子从实验室路线到可放大工艺之间存在巨大的鸿沟。
本文选取两个近年颇具代表性的分子——Enlicitide decanoate(Agenus/Bristol Myers Squibb,口服PCSK9抑制剂)和Icotrokinra(Johnson & Johnson,2026年3月获FDA批准的口服IL-23R环肽抑制剂)——梳理其公开文献与专利中披露的合成路线演进,重点关注工艺层面的关键决策:为什么选择液相而非固相?关环步骤在什么时序引入?如何通过结晶替代色谱?这些问题背后折射的,是环肽从概念到产品的工程逻辑。
1.Enlicitide decanoate
Enlicitide decanoate 是一类口服PCSK9抑制剂,其作用机制是通过阻断PCSK9与低密度脂蛋白受体(LDLR)的结合,从而抑制LDLR的降解,增加肝细胞表面LDLR数量,最终促进血液中LDL胆固醇的清除。该机制在临床上已被抗体类药物验证有效,但小分子或口服形式的PCSK9抑制剂长期难以实现,因此这一类环肽分子具有明确的药理学价值。从分子结构来看,enlicitide decanoate 属于高度复杂的环肽体系。该分子由8个氨基酸单元组成(以及2个非氨基酸构件),其中仅有2个为天然氨基酸,其余均为经过结构改造的非天然氨基酸,同时嵌入多个非肽结构片段,包括烯烃连接单元、芳香结构以及带电侧链。这些结构特征使其在本质上已经脱离传统多肽的范畴,更接近于小分子与多肽之间的杂合系。
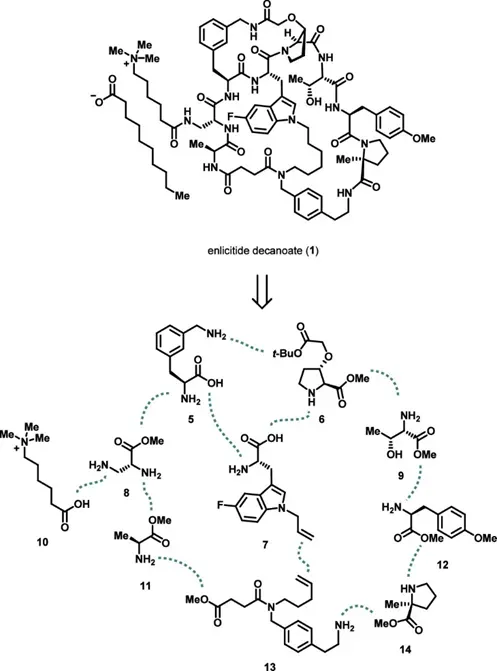
图片来源:JACS,下同
从结构上看,该分子由三个单元环共同构成一个整体的三环体系。首先,Northern fragment 内部通过分子内酰胺化形成一个17元内酰胺环,该环完全由肽键构成,属于典型的环肽结构单元;其次,在全分子组装完成后,通过另一处分子内酰胺化形成主环,该主环覆盖大部分骨架,构成37元内酰胺大环,是整个分子的核心框架;第三个环则来源于侧链之间的跨链连接,在两个烯烃片段之间通过关环复分解反应形成碳–碳双键,从而构建一个由烃链组成的桥连环结构。这种多环体系对PCSK9蛋白表面特定区域具有较强结合能力,同时提高了分子的代谢稳定性与口服生物利用度。但从合成角度看,这种结构带来了明显约束:多个关环步骤之间相互影响,任何一步构象偏差都会降低后续反应成功率。
在合成策略上,分子中非天然氨基酸比例较高,并包含烯烃连接(前体肽中的烯烃还原为烷烃)与非肽键结构,使得固相多肽合成难以适用。一方面,关环等反应难以在固相中高效进行;另一方面,多个手性中心的构建依赖溶液相或酶催化方法;同时,带正电的铵盐侧链被刻意推迟至最后一步引入,这是出于工艺可控性。一旦在早期引入带电结构,中间体的对离子需要在每一步操作中进行管理,可能对后续偶联与关环反应产生干扰。
此外,该分子的开发目标是用于心血管疾病长期治疗,其潜在患者数量巨大,对产量的需求远高于肿瘤药物或抗生素等传统复杂分子。这一背景使得路线设计必须兼顾效率与可放大性。传统依赖柱层析的纯化方式难以适用,路线中需要尽可能通过结晶、沉淀及物性调控实现纯化,并减少步骤数与物料损失。
在上述生物学需求与化学约束的共同作用下,该工作的核心问题不在于单纯完成分子构建,而在于建立一条能够在复杂结构控制、反应选择性与工业放大之间取得平衡的合成路线。
Northern fragment
Northern fragment 的改进集中在非天然氨基酸6的构建方式、Trp单元构建方式以及关环策略。
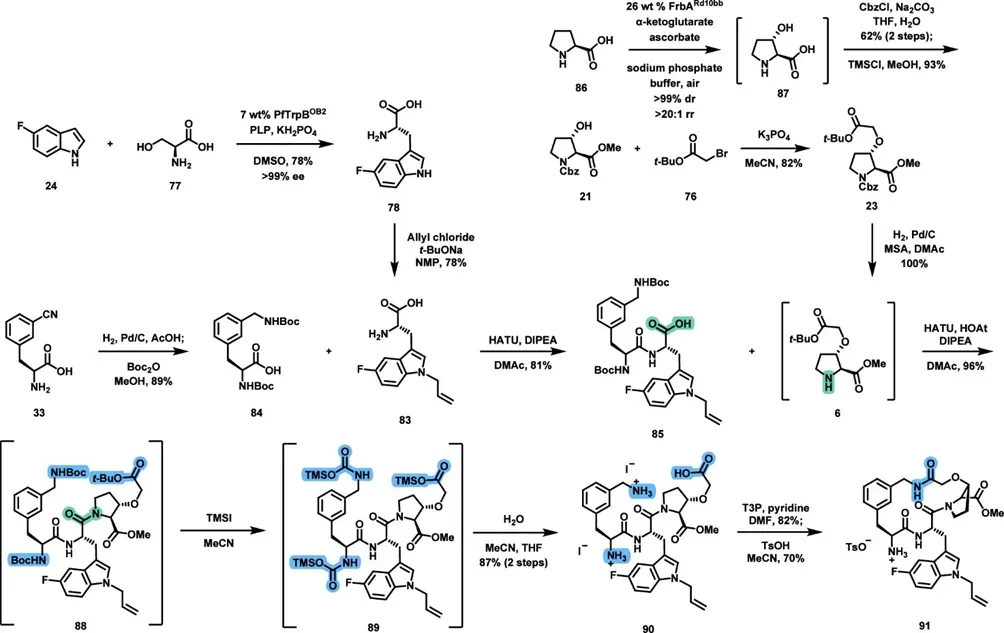
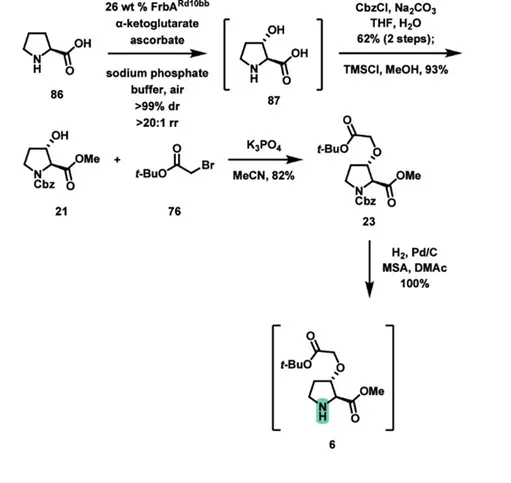
起始原料为L-proline(86)。首先在水相中使用α-酮戊二酸依赖型羟化酶变体进行C3位选择性羟化,反应直接在酶催化体系中完成,引入3-hydroxyproline结构87。该反应具有>99:1的非对映选择性,同时避免了早期路线中需要构建两个手性中心的问题。反应结束后,通过CBz保护氨基并进行羧基的酯化,得到化合物21。随后进行烷基化反应,利用tert-butyl bromoacetate 76引入tert-butyl acetate侧链制备23。之后通过Pd/C催化氢化脱除CBz保护基,得到非天然氨基酸6。
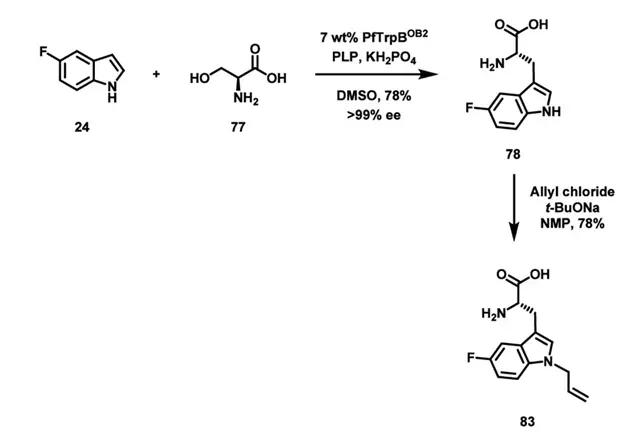
第二个关键单元83的构建采用酶催化路线。以5-fluoroindole(24)与L-serine(77)为底物,在工程化TrpB酶作用下直接偶联生成手性氨基酸78。该步骤避免了传统方法中的手性分离。随后进行酯化和保护基调整,并通过烷基化引入allyl基团,得到N-allyl-tryptophan衍生物83。值得注意的是,吲哚的allylation反应,是在Nα未被保护的情况下进行的,该反应的化学选择性相对于之前的合成路线节省了很多步骤的中间反应。
第三个单元为aminomethyl-phenylalanine(84),该单元由腈前体33经两步转化得到。首先在H₂/Pd-C、AcOH条件下将腈还原为伯氨基,得到侧链氨甲基结构;随后在MeOH中加入Boc₂O,对分子中的α-氨基和新生成的侧链氨基同时进行Boc保护,得到双Boc保护的84。
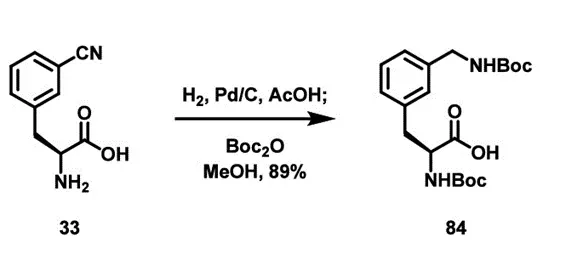
三段构建完成后进行片段拼接。首先将84的羧酸在HATU条件下活化,再加入83进行偶联。该操作顺序避免了对83羧酸进行保护。随后直接使用氢化后的6的粗反应液进行第二次偶联,得到三肽中间体88。该过程中避免中间体分离,直接串联反应。随后进行去保护并形成关环前体。在该体系中未对两个氨基进行区分保护,而是利用其pKa和位阻差异在关环步骤中实现选择性。关环采用T3P作为缩合试剂,在分子内酰胺化条件下形成宏环结构。该反应条件温和,避免外消旋。
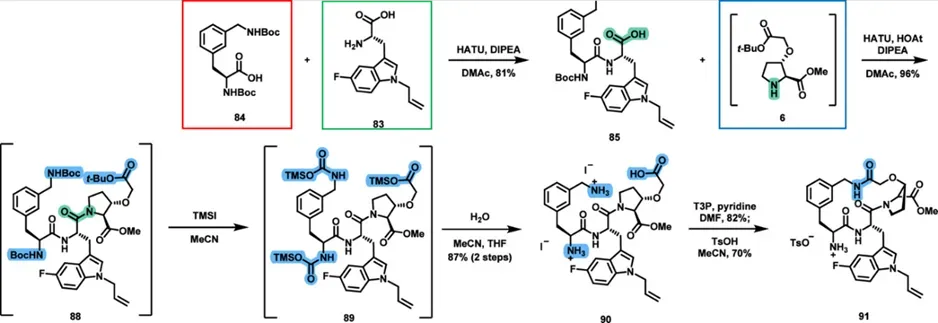
值得注意的是,环化反应前的中间体(90)可以形成晶体盐,从而实现纯化。随后经游离碱处理再进行关环反应,最后通过TsOH形成稳定的tosylate盐(91)。该步骤不仅完成结构构建,同时提供关键纯化节点。
Southern fragment
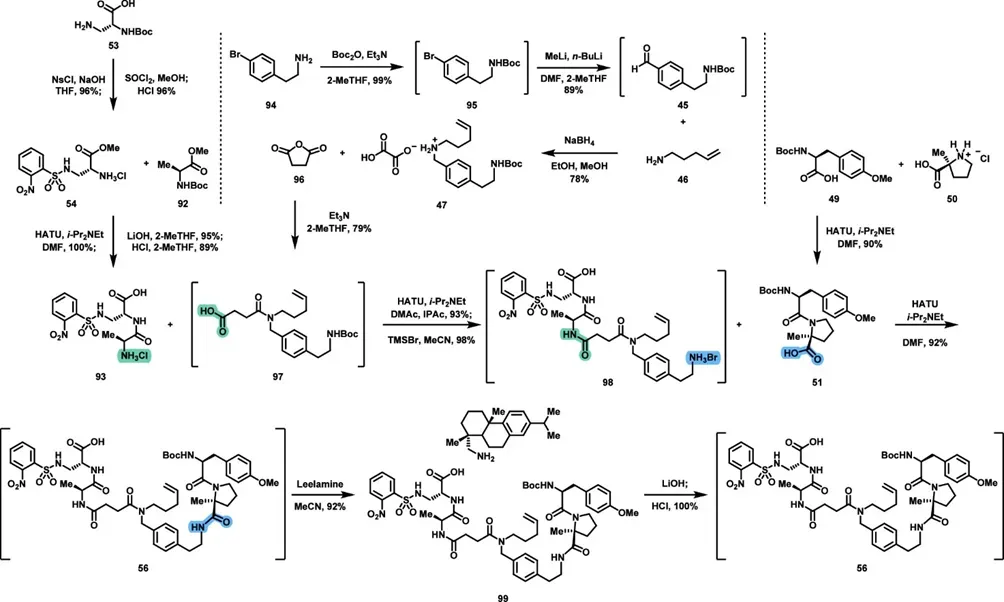
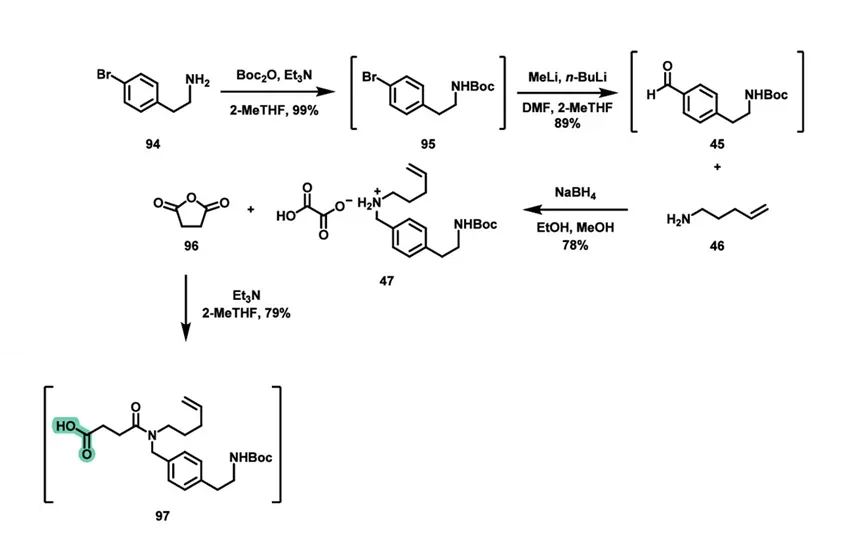
Southern fragment 的优化核心在于非肽结构片段构建方式以及结晶纯化策略。反应起始于(4-bromophenyl)ethan-1-amine(94)。首先进行Boc保护,随后通过锂卤交换反应生成芳基锂中间体,再与DMF反应实现甲酰化,得到benzaldehyde类化合物(45)。该中间体不分离,直接进入下一步。随后进行还原胺化,使用pent-4-en-1-amine(46)与醛反应,在还原条件下得到二级胺。该步骤引入烯烃链,为后续RCM(Ring-Closing Metathesis)提供反应位点。接着使用succinic anhydride进行酰化开环反应,引入羧酸功能,得到中间体97。该羧酸作为连接位点与肽段偶联。
二肽部分51由Boc-O-methyl-L-tyrosine(49)与2-methyl-L-proline(50)偶联获得。
另一部分,Boc-D-Dap-OH(53)用 2-nitrobenzenesulfonyl chloride 对侧链氨基进行保护,得到nosyl 保护的胺,再用SOCl2/MeOH进行甲酯化,得到中间体54。这里选择 nosyl 保护基并不是常规保护基替代问题,而是出于整个全合成后段的策略需要。作者在第一代路线中就说明,他们希望把带铵盐性质的侧链尽量推迟到最后一步引入,因此需要一个可以稳定经历前面多步转化、又能在后期选择性脱除的氨基保护基。54就是在这种策略下设计出来的 Southern fragment 关键构件。54之后与Boc-Ala-OMe缩合得到二肽中间体,经过LiOH介导的甲酯水解和HCl的脱Boc反应得到构件93。
第三部分,Boc-Tyr(Me)-OH (49) 和2-MePro.HCl (50) 先发生酰胺偶联,构建二肽片段51。
在Southern fragment的组装阶段,93先与97 在HATU介导下发生酰胺键形成,得到缩合产物。随后对该缩合产物用TMSBr / MeCN 处理脱Boc,得到中间体98。中间体98与二肽51在 HATU条件下再次发生酰胺偶联,得到56。56随后与 leelamine 在 MeCN 中成盐,形成可结晶分离的99,收率 92%。这一步是结晶纯化的核心。作者做了高通量结晶筛选,在数千次实验中只找到这一个有效命中,即leelamine 盐99。最后,对 99依次用 LiOH 和 HCl 处理,得到自由酸,得到最终的 Southern fragment 56。Southern fragment合成路线的关键改进在于结晶纯化,可直接获得高纯度产物。该步骤替代了原路线中的多次柱层析,是Southern fragment路线中最重要的工艺改进。
最终拼接与关环
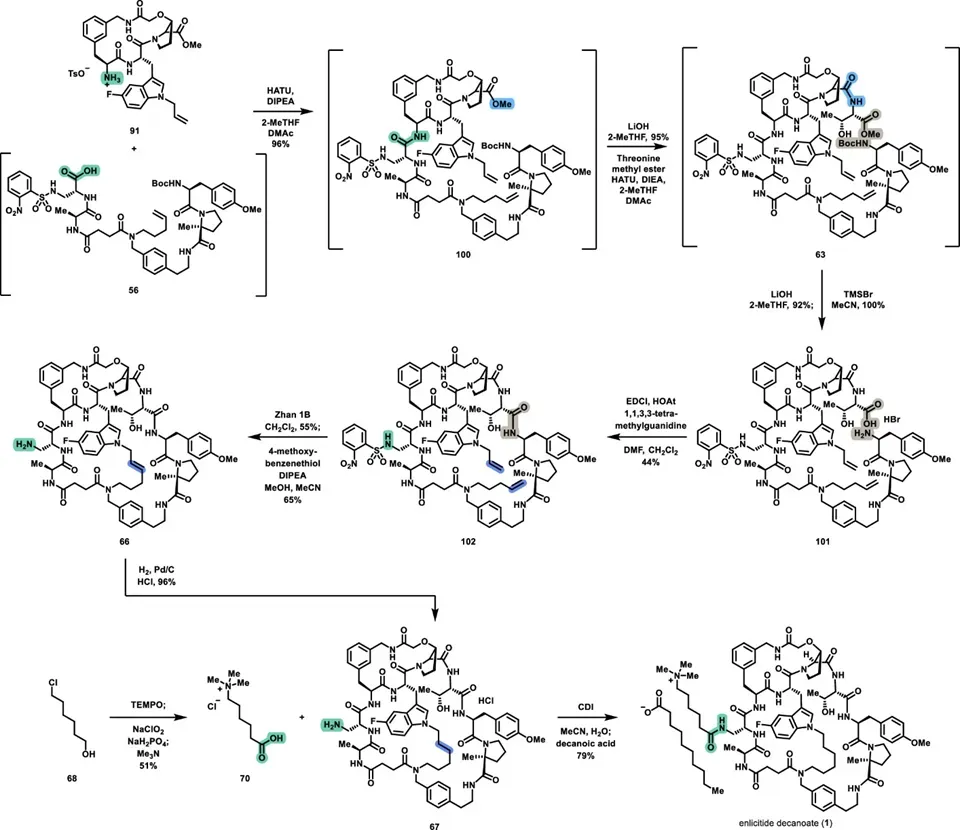
Northern fragment(91)与Southern fragment(56)首先在进行酰胺偶联,采用HATU体系完成。反应后不进行纯化,粗产物直接进入下一步。随后进行第一步皂化,水解甲酯得到羧酸,然后偶联引入L-threonine。之后进行第二次皂化,并同时脱除Boc保护基,得到完整线性前体101。随后进行环化,在分子内酰胺化条件下形成大环结构102,使用EDC/HOAt作为缩合试剂。该步骤在稀溶液条件下进行,以抑制分子间副反应。
接下来进行关键的RCM(ring-closing metathesis),使用钌催化剂完成烯烃关环,形成核心骨架。该步骤放置在后期,以降低催化剂使用量并提高整体经济性。RCM后进行氢化,使用Pd/C还原双键,形成稳定的烷基连接结构。随后进行nosyl脱保护,使用对甲氧基苯硫酚完成去保护,得到中间体67。最后引入侧链。侧链由6-chlorohexan-1-ol经连续氧化生成羧酸,再与三甲胺反应生成铵盐70。通过酰胺偶联将其连接至核心结构67,最终通过结晶得到enlicitide decanoate 1。
最终优化后的合成路线将Enlicitide decanoate 的总步数由第一代的 63 步缩短至 43 步,最长线性步骤降至 21 步,其中 Northern fragment 为 14 步,Southern fragment 为 16 步。在整体效率上,第二代路线的总收率较第一代提高约三个数量级,从约0.00007% 提升至约0.07%,尽管绝对收率仍然较低,但已显著改善物料通量。
工艺层面,通过在Southern fragment 中引入leelamine 成盐结晶中间体99,以及在 Northern fragment 中建立可结晶中间体,实现了对晚期中间体的有效分离,基本替代了色谱纯化;同时,通过调整片段拼接顺序并将RCM 等关键反应推迟至后期,降低了贵金属使用量并提高了反应成功率。整体来看,该路线在步骤数、收率、纯化方式与放大可行性之间取得了平衡,使这一高度复杂的三环宏环肽体系具备了实际工业化生产的可能。
2.Icotrokinra
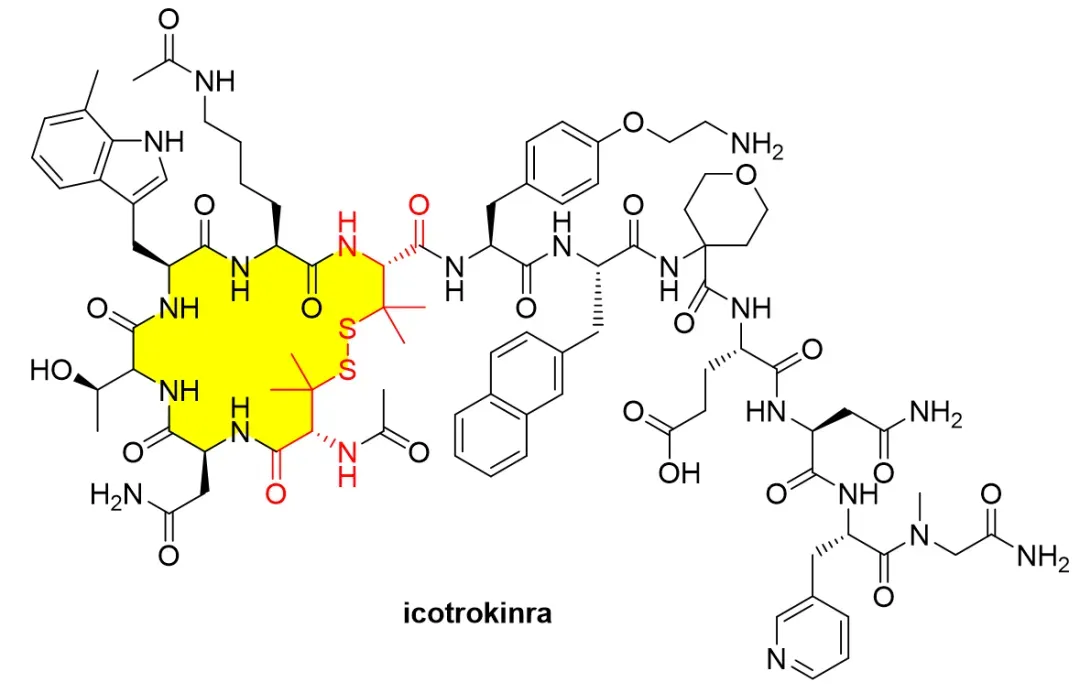
Icotrokinra 是强生开发的一类针对白细胞介素-23受体(IL-23R)的口服环肽抑制剂,属于近年来逐步受到关注的环肽类免疫调节药物,于2026年3月17日获得FDA批准,针对中重度斑块状银屑病。
Icotrokinra由13氨基酸残基组成,通过两个penicillamine (Pen,D-青霉胺)位点之间形成的二硫键构建单环结构,同时在序列中引入多种非天然氨基酸或修饰残基,例如 7-甲基色氨酸、取代苯丙氨酸、2-萘基丙氨酸以及四氢吡喃结构单元等,使其在构象稳定性、受体结合能力以及代谢稳定性方面得到显著优化。分子通常以 N 端乙酰化、C 端酰胺化的形式存在,进一步增强其体内稳定性。
在作用机制上,icotrokinra通过与IL-23 受体结合,阻断IL-23 与其受体的相互作用,从而抑制下游Th17 细胞相关炎症信号通路。这一通路在炎症性肠病、银屑病等多种自身免疫疾病中具有关键作用,因此该类环肽被开发为口服或系统给药的免疫调节候选药物。相较于抗体类IL-23 抑制剂,这类环肽在分子尺寸、组织渗透性以及潜在给药方式上具有一定差异化优势。
从化学角度看,icotrokinra兼具多肽与小分子特征,其结构中既包含典型的肽键骨架,又引入多种侧链修饰与刚性环结构,使其在保持较高结合亲和力的同时,提高了对酶降解的抵抗能力。这种高修饰度环肽的设计思路,也体现了近年来多肽药物从天然序列向高度工程化结构演进的趋势。
2.1WO2021146441A1
强生的专利WO2021146441A1给出的合成路线是最常规的固相合成,液相I2环化。
SPPS固定相为Rink Amide树脂,0.66 mmol/g。Fmoc脱除条件为用20% 4-甲基哌啶 / DMF,分两次处理树脂,第一次3–5 min,第二次20–30 min,然后DMF 洗 3 次。氨基酸缩合条件为,将3.0 equivFmoc-氨基酸与4.5 equiv Oxyma溶于DMF,先加入3.9 equiv DIC预活化 15 min,然后加到已脱 Fmoc 的树脂上;再在反应约 15 min 后补加第二份 DIC (2.6 equiv)。反应进程用 Kaiser test 监测,完成后 DMF 洗 3 次,进入下一轮脱保护/缩合循环。最终的全保护多肽序列为Pen(Trt)-Asn(Trt)-Thr(tBu)-7MeTrp-Lys(Ac)-Pen(Trt)-AEF-Nal-THP-Glu(OtBu)-Asn(Trt)-3Pal-Sar-amide resin。
最后一轮Fmoc 脱除后,加入乙酰化试剂THF / 乙酸酐 / 吡啶 = 80:10:10,震荡 30 min。随后树脂 DMF 洗 3 次,再 DCM 洗 5 次,并在真空下 1.5 h,然后进入裂解。
切割和去保护反应条件为:TFA / 水 / TIPS / DODT (3,6-dioxa-1,8-octanedithiol) = 90:5:2.5:2.5 的裂解液,加入到装有树脂的管中,震荡 2 h。之后过滤除树脂,把滤液分装,用冷乙醚沉淀,离心,倾去乙醚,再做 2 次乙醚洗,得到粗还原态线性肽。
液相环化条件为:把粗肽溶于20% ACN / 水,浓度大约 2 mg/mL;在搅拌下逐滴加入乙酸中的饱和碘溶液,直到体系保持黄色/棕色不再褪去。反应完成后,用分析 HPLC 和 LCMS 监测;随后加入固体抗坏血酸把多余碘淬灭到溶液澄清,再上反相 HPLC(C18)纯化。
2.2WO 2024/110477 A2
强生的另一篇专利WO 2024/110477 A2中采取了液相合成的手段制备icotrokinra。
其整体合成路线是将icotrokinra分成两个大的片段,一个为C端直链肽,另一个为N端环肽,这两部分都在液相中合成,然后再在液相中进行片段缩合,制备全保护的icotrokinra,最后进行global deprotection获得icotrokinra。
线性片段从 C 端开始构建。以 1.0 equiv. Boc-3Pal-OH 为起点,在 1.2 equiv. H-Sar-NH₂、1.2 equiv. PivCl、2.0 equiv. NMM 下,于THF 中低温(0–5 °C)活化后升至室温反应,完成缩合。随后用TFA / DCM(1:1)处理(室温,约 1 h)脱除 Boc,浓缩后直接进入下一步。
得到的二肽中间体与1.2 equiv. Z-Asn-OSu 在 DMF 中于室温反应,完成后用 H₂(1 atm)/ 10 wt% Pd/C / MeOH 氢解(室温,2–4 h)去除Z。随后与 1.2 equiv. Z-Glu(OtBu)-OSu 在 DMF 中继续偶联,反应结束后再次在 H₂ / Pd-C / MeOH 条件下脱Z,得到中间体四肽。
接着将该片段与1.2 equiv. 二肽原料Fmoc-2Nal-THPGly-OH在 1.2 equiv PyAOP、2.0 equiv DIPEA、DMF条件下于室温反应(一般2–6 h),完成偶联。之后用DBU / DMF(约20–30%)室温处理(10–30 min)去除 Fmoc,得到最终线性片段H-[8–13]-NH₂。
环肽片段的构建,是通过将1.0 equiv. Ac-Pen(Trt)-Asn-Thr(tBu)-OH与 1.0–1.2 equiv. H-7MeTrp-Lys(Ac)-Pen(Acm)-Tyr(2-Boc-ea)-OMe 在 1.2 equiv DIC, 1.2 equiv Oxyma, DMF 中于室温反应(2–12 h)完成缩合,得到线性七肽。这两个三肽和四肽原料的合成也是在也是在液相中进行的,过程分别如下:
Ac-Pen(Trt)-Asn-Thr(tBu)-OH的制备路线:
先由 1.0 eq Z-Asn-Thr(tBu)-OMe在 2-MeTHF 中进行氢解,加入 10 wt% 湿 Pd/C,在 40 psi H2、24–27 °C 下反应 4 h,过滤除催化剂后,再加入 1.0 eq 对甲苯磺酸的 2-MeTHF 溶液,得到 H-Asn-Thr(tBu)-OMe 的溶液。
接着以此为底物,与1.0 eq Fmoc-Pen(Trt)-OH、1.07 eq TBTU 以及 2.0 eq DIPEA 反应,体系为 DMSO / EtOAc,在 20–25 °C 下偶联,约 1 h 完成,之后经 NaHCO3水洗、水洗、浓缩、EtOAc/庚烷沉淀,得到Fmoc-Pen(Trt)-Asn-Thr(tBu)-OMe。
再往下,该中间体经过DBU 脱Fmoc,得到H-Pen(Trt)-Asn-Thr(tBu)-OMe;随后用 Ac2O 乙酰化得到 Ac-Pen(Trt)-Asn-Thr(tBu)-OMe;最后再用 1.4 eq LiOH·H2O 在 THF / H2O、5 °C 下水解甲酯,约 5 h 后几乎反应完全,酸化到 pH 4.7,萃取、浓缩并用 EtOAc/庚烷沉淀,得到 Ac-Pen(Trt)-Asn-Thr(tBu)-OH。
H-7Me-Trp-Lys(Ac)-Pen(Acm)-Tyr(2-Boc-ea)-OMe的制备路线:
以 1.0 eq Fmoc-Pen(Acm)-OH和 1.05 eq H-Tyr(2-Boc-ea)-OMe 为底物,在 MeCN 中、0 °C 下加入0.5 eq NMM、0.5 eq Oxyma Pure,再分批加入1.1 eq EDCI,于0–5 °C 下反应16–20 h,得到Fmoc-Pen(Acm)-Tyr(2-Boc-ea)-OMe;之后在 MeCN 中、0 °C 下加入4.0 eq 十二烷基硫醇和 0.95 eq DBU,反应 10 h,经 heptane、EtOAc 和盐水体系洗涤后得到 H-Pen(Acm)-Tyr(2-Boc-ea)-OMe。
接下来向该中间体溶液中加入0.96 eq Fmoc-Lys(Ac)-OH,在 0 °C 下再加入0.5 eq NMM、0.5 eq ethyl cyanoglyoxylate-2-oxime 和 1.1 eq EDCI,反应16–20 h,经碳酸氢钠、氯化铵和氯化钠洗涤后得到Fmoc-Lys(Ac)-Pen(Acm)-Tyr(2-Boc-ea)-OMe 的溶液。
下一步脱Fmoc:以 1.0 eq Fmoc-Lys(Ac)-Pen(Acm)-Tyr(2-Boc-ea)-OMe 为底物,在 MeCN、20 °C 下加入1.5 eq 二乙胺和 3.0 eq 十二烷基硫醇,反应 7–10 h,经 heptane 洗涤、浓缩并换入 2-MeTHF,得到H-Lys(Ac)-Pen(Acm)-Tyr(2-Boc-ea)-OMe。
向该中间体溶液中加入0.97 eq Fmoc-Trp(7Me)-OH、0.5 eq ethyl cyanoglyoxylate-2-oxime 和 1.3 eq EDCI,在 0–5 °C 下反应 10 h;后处理时依次用5% NaHCO3、NH4Cl、10% NaCl 洗涤,再反复与2-MeTHF 置换至 KF含水量<0.3%,降温到 5 °C 后加晶种,搅拌 20 h,再缓慢加入10.0 vol MTBE 诱导结晶,得到Fmoc-Trp(7Me)-Lys(Ac)-Pen(Acm)-Tyr(2-Boc-ea)-OMe。最后再在 EtOAc/DMSO = 8:2 中加入 0.5 eq DBU,室温反应 1.5 h,经 5% NaHCO3 和水洗、EtOAc/MeTHF处理、IPE重结晶,得到H-7Me-Trp-Lys(Ac)-Pen(Acm)-Tyr(2-Boc-ea)-OMe。
Ac-Pen(Trt)-Asn-Thr(tBu)-Trp(7Me)-Lys(Ac)-Pen(Acm)-Tyr(2-Boc-ea)-OMe水解
将 1.0 eq Ac-Pen(Trt)-Asn-Thr(tBu)-Trp(7Me)-Lys(Ac)-Pen(Acm)-Tyr(2-Boc-ea)-OMe 溶于 THF/H2O,再加入水,在 0–10 °C 下滴加 1.5 eq NaOH 水溶液,滴加时间 30 min,然后继续反应 5 h。反应后加入 2 M HCl,升温到 15–25 °C,再加 NaCl 分相;有机相减压浓缩后,用 10% water/MeCN 连续置换溶剂,得到Ac-Pen(Trt)-Asn-Thr(tBu)-Trp(7Me)-Lys(Ac)-Pen(Acm)-Tyr(2-Boc-ea)-OH 的H2O/ACN溶液,收率 97.3%。
Ac-Pen(Trt)-Asn-Thr(tBu)-Trp(7Me)-Lys(Ac)-Pen(Acm)-Tyr(2-Boc-ea)-OH环化
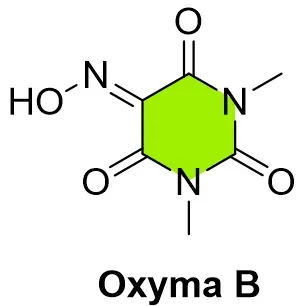
先把上一步得到的线性多肽Ac-Pen(Trt)-Asn-Thr(tBu)-Trp(7Me)-Lys(Ac)-Pen(Acm)-Tyr(2-Boc-ea)-OH与 2.25 eq 2,6-dimethylpyridine 和少量水混合;另一釜中预先配好 0.9 eq I2、2.25 eq KI、0.9 eq formic acid 的MeCN/H2O 溶液,温度控制在 20–30 °C。然后把线性肽溶液在 10.5 h 内滴加到含碘体系中,滴完后再搅拌 2 h 完成环化,得到二硫键环化的Ac-Pen*-Asn-Thr(tBu)-Trp(7Me)-Lys(Ac)-Pen*-Tyr(2-Boc-ea)-OH多肽(*代表二硫键环化桥头残基,下同)。
片段缩合
环肽Ac-Pen*-Asn-Thr(tBu)-Trp(7Me)-Lys(Ac)-Pen*-Tyr(2-Boc-ea)-OH与线性片段H-2Nal-Gly(THP)-Glu(tBu)-Asn-3Pal-Sar-NH2进行偶联。将 1.0 eq Ac-Pen*-Asn-Thr(tBu)-Trp(7Me)-Lys(Ac)-Pen-Tyr(2-Boc-ea)-OH与 1.05 eq H-2Nal-Gly(THP)-Glu(tBu)-Asn-3Pal-Sar-NH2溶于 THF,在 20–25 °C 下加入 0.5 eq Oxyma B,再在 30 min 内滴加 1.5 eq DIC,继续反应 5 h。反应后依次用 2 M HCl、5% NaHCO3水溶液和 5% NaCl 水溶液洗涤,再加入 2-MeTHF 分相,得到全保护icotrokinra多肽。
Global Deprotection
将 1.0 eq 保护icotrokinra 悬浮于 iPrOAc 中,在 15 °C 下快速加入 3 M HCl/iPrOAc,先在 15 °C 搅拌 5 min,然后在 20 min 内升到 30 °C,继续反应 5 h。反应后降到 -5 °C,加入约 5% NaOH 水溶液,再补加水,把体系调到 pH 2.8,分相后取水相,再继续把 pH 调到 4.25,搅拌 1 h 形成白色悬浊液;然后再把 pH 调到 4.50,于 20 °C 下搅拌 14 h,过滤、水洗,在 45 °C 真空干燥 24 h,得到icotrokinra,收率 89%,HPLC 纯度 95%。
对比两条路线,可以归纳出几个共性规律:关环步骤的时序是效率的核心变量;结晶纯化对规模化生产的重要性往往超过反应本身的优化;酶催化与非天然氨基酸的引入已成为复杂多肽工艺的常规手段。两者的差异同样值得关注——Enlicitide三环骨架带来的合成复杂度,使其第二代路线历经大幅优化后总步数仍达43步;Icotrokinra凭借二硫键关环的相对简洁性,global deprotection收率可达89%。这一对比提示,分子设计阶段对可合成性的考量,对工艺开发的影响是根本性的。
从更宏观的视角看,这两个案例都指向同一个趋势:环肽正在从"合成化学家的挑战题"逐步演变为具备工业生产基础的药物类别。随着酶催化、流动化学与结晶工程在多肽工艺中的应用日趋成熟,这条从结构复杂性到临床可及性的路径,正在变得越来越清晰。
Ref.
Li, H. et al. Total Synthesis of Enlicitide Decanoate. J. Am. Chem. Soc. 2025, 147, 11036−11048.
Bruno, P. et al. WO 2024/110477 A2. JANSSEN PHARMACEUTICA NV. International Publication Date 30 May 2024 (30.05.2024)
Sun, C. et al. WO2021146441A1 Peptide inhibitors of interleukin-23 receptor and their use to treat inflammatory diseases. Janssen Biotech Inc Protagonist Therapeutics Inc. International Publication Date 22 July 2021 (22.07.2021)