


5. 乳腺癌与人体免疫系统的关系
乳腺癌的发生与进展并非单纯的肿瘤细胞自主增殖过程,而是肿瘤细胞与宿主免疫系统持续互作、共同演化的动态过程。从免疫监视到免疫逃逸,从”冷肿瘤”微环境到免疫检查点抑制剂(Immune Checkpoint Inhibitor, ICI)治疗响应,免疫系统在乳腺癌的全程管理中扮演着决定性角色。本章将从肿瘤免疫编辑、肿瘤微环境(Tumor Microenvironment, TME)免疫景观、免疫治疗耐药机制以及免疫系统与治疗策略的整合四个维度,系统阐述当前研究进展。
5.1 肿瘤免疫编辑与免疫逃逸
5.1.1 免疫编辑三阶段
肿瘤免疫编辑(Cancer Immunoediting)理论由Schreiber等提出,描述了免疫系统与肿瘤从起始到临床显现的全程互作,包括消除(Elimination)、平衡(Equilibrium)和逃逸(Escape)三个阶段。该理论在乳腺癌中已获得了直接的基因组学证据支持。2024年8月,Zhang等人在Nature Immunology发表的研究使用乳腺癌基因工程小鼠模型(Genetically Engineered Mouse Model, GEMM)进行单细胞RNA测序(single-cell RNA sequencing, scRNA-seq),无偏地绘制了从早期到晚期乳腺肿瘤的免疫编辑全图谱。研究发现,晚期肿瘤中先天性和适应性免疫基因被显著抑制,而调控细胞内在恶性程度的基因大多未被编辑——这一发现直接证实免疫编辑主要作用于免疫相关基因而非促癌基因。值得注意的是,干扰素刺激基因(Interferon-Stimulated Genes, ISGs)构成了下调肿瘤基因的主导列表,先天免疫基因的抑制尤为突出。该研究的意义在于,首次在乳腺癌模型中区分了”免疫编辑”与”基因突变驱动的肿瘤进化”这两个并行过程,提示肿瘤逃逸免疫控制的方式主要通过表观遗传重编程而非基因突变积累。
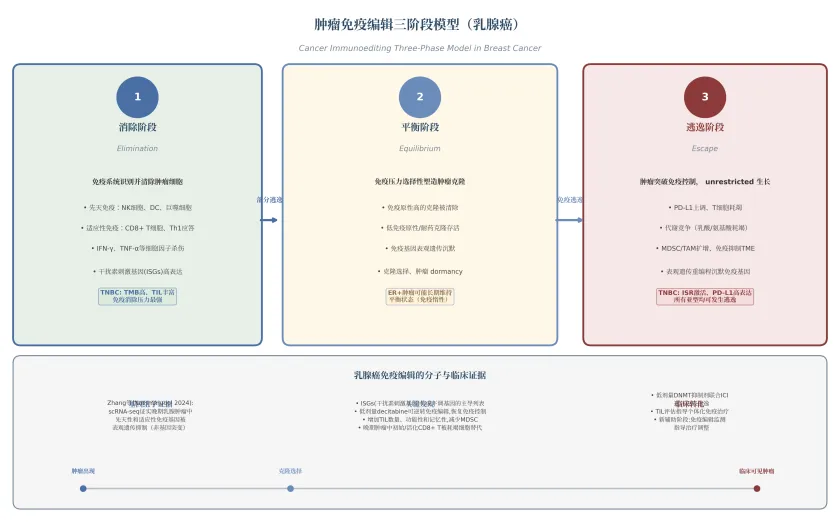
图5-2 肿瘤免疫编辑三阶段模型
在消除阶段,三阴性乳腺癌(Triple-Negative Breast Cancer, TNBC)由于较高的肿瘤突变负荷(Tumor Mutational Burden, TMB)和更频繁的肿瘤浸润淋巴细胞(Tumor-Infiltrating Lymphocytes, TILs),呈现更为活跃的免疫消除反应。平衡阶段的特点是免疫压力对肿瘤克隆进行选择性筛选,免疫原性高的克隆被清除,低免疫原性或获得耐药机制的克隆存活并扩增。ER阳性(Estrogen Receptor-positive, ER+)乳腺癌因免疫原性相对较弱,可能长期维持平衡状态而表现为”免疫惰性”。逃逸阶段则是肿瘤细胞突破免疫控制、实现不受限制生长的阶段,涉及PD-L1上调、T细胞耗竭、代谢竞争和免疫抑制微环境构建等多重机制。上述三阶段的转化机制与临床特征总结于图2。
5.1.2 表观遗传驱动的免疫编辑
Zhang等人的研究揭示了一个关键机制:免疫编辑的核心驱动力是表观遗传抑制而非基因突变。在晚期乳腺肿瘤中,研究者观察到免疫基因启动子区域DNA甲基化水平显著升高,导致ISGs、抗原呈递 machinery组分和趋化因子基因的表达沉默。更具转化意义的是,低剂量地西他滨(decitabine,一种DNA甲基转移酶抑制剂)可逆转这种免疫编辑效应——恢复免疫控制、增加肿瘤浸润淋巴细胞的数量与功能性、减少髓系来源抑制细胞(Myeloid-Derived Suppressor Cells, MDSCs)的积累,并赋予T细胞记忆性表型。这一发现为”表观遗传-免疫联合治疗”策略提供了直接的临床前证据,提示低剂量去甲基化药物可能将免疫”冷”肿瘤转化为”热”肿瘤。然而,需要强调的是,decitabine的最佳免疫调节剂量仍需在临床中精确界定,因为细胞毒性剂量可能杀伤活化的淋巴细胞,而低剂量的安全性和有效性窗口在人体中的验证尚待完成。
5.1.3 乳腺癌免疫逃逸核心机制
乳腺癌免疫逃逸是一个多因素协同的网络化过程。如图1所示,逃逸机制主要涉及四个维度:PD-L1/免疫检查点通路上调、代谢竞争、MDSCs介导的免疫抑制以及肿瘤相关巨噬细胞(Tumor-Associated Macrophages, TAMs)的M2样极化。
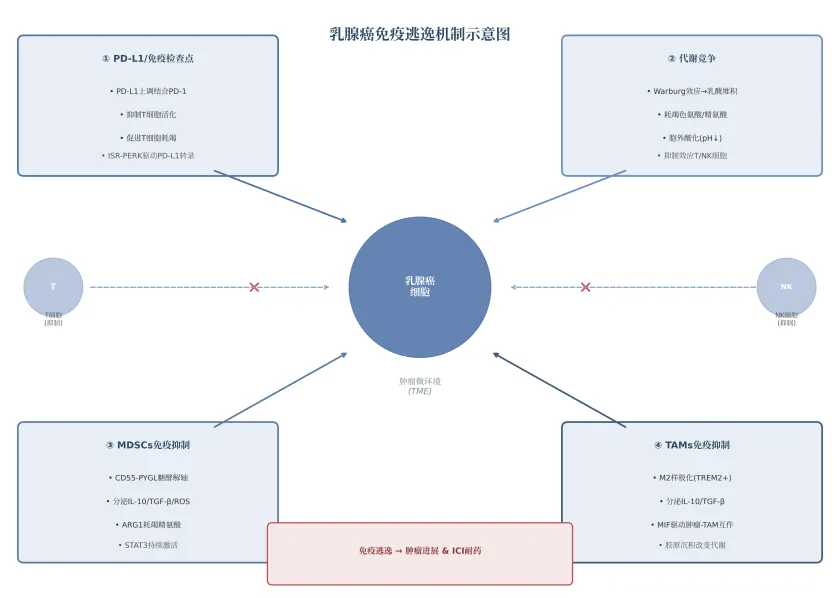
图5-1 乳腺癌免疫逃逸机制示意图
在PD-L1/免疫检查点维度,整合应激反应(Integrated Stress Response, ISR)通路中的PERK激酶可通过磷酸化eIF2α激活ATF4转录因子,直接驱动PD-L1基因转录。代谢竞争维度方面,TNBC的高糖酵解活性产生大量乳酸,通过单羧酸转运蛋白(Monocarboxylate Transporters, MCTs)释放至胞外,造成TME酸化并抑制效应T细胞和NK细胞的功能。MDSCs维度上,CD55被发现为MDSCs上广泛过表达的保守表面标志物,通过CD55-糖原磷酸化酶(PYGL)轴驱动MDSCs糖酵解并维持STAT3激活,遗传敲除CD55可显著增强抗PD-1治疗效果。TAMs维度上,TREM2+巨噬细胞与PD-1+淋巴细胞形成的免疫抑制微环境空间共定位与不良预后显著相关。这些逃逸机制并非独立运作,而是相互强化——例如,ISR通路同时驱动PD-L1上调和代谢重编程,CAF-TAM轴形成免疫抑制网络,共同构建了一个多维度的免疫屏障。
5.2 肿瘤微环境免疫景观
5.2.1 TNBC TME特征:9种预后生态型
Wu等人于2021年在Nature Genetics发表的研究通过整合scRNA-seq和空间转录组学技术,在乳腺肿瘤中鉴定出9种主要细胞类型和9种预后生态型(ecotypes),由细胞共定位模式定义而非单纯的细胞丰度。这一发现突破了传统”热肿瘤/冷肿瘤”二元分类的局限,揭示了TME的空间异质性是决定预后的关键因素。核心发现包括:基底样肿瘤中存在互斥空间区域——上皮间质转化(Epithelial-Mesenchymal Transition, EMT)富集的浸润前沿与增殖性核心形成鲜明对比;32%的管腔样(luminal)肿瘤携带基底/HER2样亚克隆,可能驱动治疗耐药;代谢梯度沿肿瘤-间质轴呈规律性分布——肿瘤边缘脂质代谢上调,远端间质以氧化磷酸化为主。这一多维空间框架为超越传统免疫分类的精准肿瘤学提供了新的理论基础,尽管其临床转化仍需前瞻性验证。
5.2.2 肿瘤相关巨噬细胞(TAMs)
TAMs是乳腺癌TME中最主要的免疫抑制细胞群体。在TNBC中,TAMs通过分泌IL-10和TGF-β、诱导胶原沉积改变代谢环境、表达IRF8转录因子等途径诱导T细胞耗竭。空间转录组学研究进一步揭示,MIF(巨噬细胞迁移抑制因子)介导的空间限制性肿瘤-巨噬细胞互作促进M2样极化,建立免疫抑制微环境。FAP+癌症相关成纤维细胞(Cancer-Associated Fibroblasts, CAFs)通过纤维连接蛋白1-整合素α5β1轴诱导巨噬细胞M2样极化,而AKAP12+成纤维细胞亚群则通过促进TAM的M2极化决定TNBC免疫抑制微环境的形成。针对TAMs的治疗策略面临挑战:CSF1R抑制剂(如pexidartinib)虽在腱鞘巨细胞瘤中获批,但在乳腺癌GEMM模型中,BLZ945虽有效减少了TAM数量,却对肿瘤负荷、进展或生存无显著影响——提示非经典单核细胞的代偿性增加可能限制了CSF1R单靶点抑制的疗效。
5.2.3 MDSCs与免疫抑制
MDSCs在乳腺癌中具有亚型特异性异质性。TNBC更易出现显著的多形核MDSC(PMN-MDSC)积累,伴随CXCR2轴(CXCL8/IL-8)超活化和缺氧驱动的免疫抑制极化;HER2+和HR+肿瘤虽基线MDSC浸润较低,但在治疗压力下仍可显著扩增。CD55+ MDSCs是近年发现的关键免疫逃逸驱动因素:CD55通过CD55-PYGL轴维持MDSCs的糖酵解代谢和STAT3激活,药理学抑制剂tomatidine可破坏该相互作用,显著改善抗PD-1治疗效果。此外,MDSC中的ISR激活(通过PERK/GCN2轴)进一步抑制抗肿瘤免疫,形成代谢-免疫交叉调控网络。
5.2.4 癌症相关成纤维细胞(CAFs)
CAFs在乳腺癌TME中表现出显著的可塑性。研究鉴定出3-5种可塑性状态的空间分离:TGF-β-myCAF与TREM2+ TAM的空间共定位形成免疫抑制生态型,与不良预后相关;而iCAF(炎症性CAF)与CD8+ T细胞的共定位则促进免疫抑制效应。CAF还通过物理屏障效应——胶原沉积和基质重塑——阻碍免疫细胞向肿瘤核心的浸润。Detox-iCAF可通过DPP4和YAP1/TEAD信号通路转化为其他亚型,提示CAF状态具有动态可逆性。靶向CAF的治疗策略(如FAP靶向抗体、TGF-β抑制剂)与ICI联合正在积极探索中,但CAF的异质性和可塑性为精准干预带来挑战。
表1 乳腺癌肿瘤微环境主要免疫细胞组成与功能
细胞类型 | 主要亚群 | 在乳腺癌TME中的分布特征 | 核心功能 | 与预后的关系 |
TILs(肿瘤浸润淋巴细胞) | CD8+ T细胞、CD4+ T细胞 | TNBC和HER2+亚型浸润丰富;ER+亚型相对较少 | 抗肿瘤免疫的核心效应细胞;TILs≥10%与pCR率显著相关 | 高TILs与TNBC/HER2+患者BCFI和OS改善显著相关 |
TAMs(肿瘤相关巨噬细胞) | M1样(促炎)、M2样(免疫抑制,TREM2+) | 在所有亚型中均为最丰富的髓系细胞;TNBC中M2样极化更显著 | 分泌IL-10/TGF-β抑制T细胞;表达IRF8诱导T细胞耗竭 | TREM2+ TAM与PD-1+淋巴细胞共定位与不良预后相关 |
MDSCs(髓系来源抑制细胞) | PMN-MDSC、M-MDSC | TNBC中PMN-MDSC积累最显著;治疗压力下各亚型均可扩增 | CD55-PYGL轴维持糖酵解和STAT3激活;分泌ROS/ARG1 | 高MDSC浸润与免疫治疗耐药和不良预后相关 |
CAFs(癌症相关成纤维细胞) | myCAF、iCAF、Detox-iCAF | 基底样肿瘤中FAP+CAF丰富;3-5种状态空间分离 | 胶原沉积形成物理屏障;诱导TAM M2极化 | FAP+CAF与TREM2+TAM共定位生态型预后最差 |
NK细胞 | 成熟NK、脱颗粒NK | 功能受TME酸化和乳酸积累抑制 | 直接杀伤肿瘤细胞;ADCC效应(尤其HER2+亚型) | 低NK活性与免疫逃逸和进展相关 |
树突状细胞(DCs) | cDC1、cDC2、pDC | cDC1负责交叉呈递肿瘤抗原;pDC功能受TME酸化抑制 | 抗原呈递启动适应性免疫;cDC1与CD8+ T细胞招募相关 | 成熟DC浸润与ICI响应正相关 |
上表系统梳理了乳腺癌TME中六种关键免疫细胞群体的亚群构成、分布特征、功能机制及预后相关性。从表中可以观察到几个重要规律:第一,TNBC在所有亚型中呈现最为显著的免疫抑制TME特征——TILs丰富但伴随高度MDSC浸润和M2样TAM极化,形成”免疫激活-免疫抑制并存”的矛盾状态;第二,空间共定位模式(如TREM2+ TAM与PD-1+淋巴细胞、FAP+ CAF与TREM2+ TAM)比单纯的细胞丰度更能预测预后;第三,CAF-TAM-MDSC三者形成免疫抑制网络而非独立运作,提示联合靶向策略可能比单靶点干预更有效。这些数据为理解乳腺癌免疫治疗的响应差异提供了细胞水平的解释框架。
5.3 免疫治疗耐药机制
乳腺癌免疫治疗耐药的机制涉及代谢重编程、信号通路异常激活、DNA损伤修复增强和表观遗传修饰等多层面交叉网络。理解这些耐药机制是克服ICI治疗失败、开发联合策略的基础。
5.3.1 整合应激反应(ISR)通路
ISR通路是近年来发现的免疫治疗耐药核心代谢-免疫调控轴。该通路通过四个上游激酶——PERK、GCN2、PKR和HRI——汇聚磷酸化eIF2α(p-eIF2α),激活ATF4驱动的转录程序,构建免疫抑制TME。具体机制包括:肿瘤固有PD-L1上调、免疫抑制外泌体分泌、代谢竞争耗竭关键氨基酸(色氨酸、精氨酸)以及直接损伤T细胞抗原受体信号传导。在MDSC和DC中,ISR激活进一步抑制抗肿瘤免疫——GCN2感知氨基酸缺乏,促进Treg分化和MDSC活化;PERK-eIF2α-ATF4轴驱动PD-L1转录和免疫抑制外泌体分泌。临床前研究表明,PERK抑制剂(GSK2606414、AMG-44)、GCN2抑制剂(GCN2iB)和ISRIB(p-eIF2α下游抑制剂)与ICI联合可逆转免疫抑制。值得注意的是,这些药物尚处于早期临床开发阶段,其在人体中的安全性和最佳联合方案仍需验证。
5.3.2 代谢耐药
TNBC的高糖酵解活性(Warburg效应)产生大量乳酸,通过MCT1/2转运出细胞并由GPR81受体感知,导致TME酸化(pH降低)。乳酸化环境直接抑制效应T细胞和NK细胞的细胞毒功能,同时促进Treg存活和功能维持。LDHA高表达与MCT4高表达是TNBC预后不良的标志物。除乳酸代谢外,IDO(吲哚胺2,3-双加氧酶)介导的色氨酸耗竭和Treg生成是另一条重要的代谢耐药通路。针对代谢耐药的治疗策略包括LDHA抑制剂(FX11、gossypol)、MCT1抑制剂(AZD3965)和二甲双胍等系统降糖药物——后者可通过降低肿瘤缺氧增强PD-1抑制剂疗效,但相关临床数据尚不成熟。
5.3.3 DNA修复增强
BRCA野生型TNBC保持完整的同源重组(Homologous Recombination, HR)修复能力,不仅赋予化疗耐药性,还通过减少突变积累和新抗原产生降低肿瘤免疫原性,从而导致ICI内在耐药。相反,BRCA突变或同源重组缺陷(HR Deficiency, HRD)肿瘤因累积更多突变而呈现更高免疫原性,解释了PARP抑制剂与ICI联合在BRCA突变肿瘤中观察到的协同效应(ORR达41%-69.2%)。
5.3.4 STAT3持续激活
STAT3信号通路在TNBC中持续激活,通过多重机制维持免疫抑制TME:诱导IL-10等免疫抑制细胞因子分泌、直接上调PD-L1表达、促进Treg扩增和MDSC募集,以及抑制Th1型免疫应答。STAT3的持续激活与JAK-STAT通路的上游异常(如细胞因子过度分泌)和负调控因子(如SOCS蛋白)的功能缺失密切相关。靶向STAT3的策略(如STAT3抑制剂、JAK抑制剂)与ICI联合正在早期临床探索中,但STAT3作为转录因子的”不可成药性”历史挑战需要新型靶向技术(如PROTAC、寡核苷酸药物)的突破。
5.3.5 表观遗传耐药
表观遗传调控在乳腺癌免疫治疗耐药中发挥三重作用机制。EZH2(Enhancer of Zeste Homolog 2)介导的H3K27me3修饰沉默Th1型趋化因子CXCL9/CXCL10和肿瘤抑制基因GREB1,EZH2高表达预测TNBC不良总生存期。EZH2抑制剂(如tazemetostat)可上调MHC-I抗原呈递并招募CD8+ T细胞,与durvalumab的联合试验正在进行中(NCT04705818)。DNMT介导的DNA甲基化直接抑制免疫基因——包括MHC-I和抗原加工呈递(Antigen Processing and Presentation, APM)组分,DNMT抑制剂可激活内源性逆转录病毒诱导dsRNA/STING/IFN-I轴,恢复免疫原性。HDAC介导的组蛋白去乙酰化维持免疫抑制微环境,HDAC1抑制联合抗PD-1在乳腺癌模型中可阻止T细胞耗竭。
表2 免疫治疗耐药机制与克服策略
耐药机制 | 核心分子/通路 | 对免疫治疗的影响 | 克服策略(在研) | 证据等级 |
ISR通路激活 | PERK/GCN2/PKR/HRI→p-eIF2α→ATF4 | PD-L1上调、免疫抑制外泌体、氨基酸耗竭、T细胞功能受损 | PERK抑制剂(AMG-44)、GCN2iB、ISRIB联合ICI | 临床前 |
代谢重编程(Warburg效应) | LDHA/MCT↑、乳酸堆积、TME酸化 | 抑制效应T/NK细胞功能、促进Treg存活、GPR81介导免疫抑制 | LDHA抑制剂(FX11)、MCT1抑制剂(AZD3965)、二甲双胍联合ICI | 临床前/早期临床 |
DNA修复增强 | BRCA wt/HR完整 | 新抗原产生减少、肿瘤免疫原性降低 | PARP抑制剂+ICI(BRCA突变人群获益) | I-II期临床 |
STAT3持续激活 | JAK→STAT3→IL-10/PD-L1/Treg | 免疫抑制细胞因子分泌、PD-L1上调、MDSC募集 | STAT3抑制剂、JAK抑制剂联合ICI | 早期临床 |
EZH2介导H3K27me3 | EZH2/PRC2→CXCL9/10沉默、MHC-I↓ | Th1趋化因子沉默、抗原呈递抑制、CD8+ T细胞招募受阻 | Tazemetostat(EZH2抑制剂)+ durvalumab(NCT04705818) | I-II期临床 |
DNMT介导DNA甲基化 | DNMT1/3B→MHC-I/APM基因沉默 | 免疫基因表观遗传沉默、ERVs抑制 | 低剂量decitabine/azacitidine+ICI(NCT02811497) | I-II期临床 |
HDAC介导去乙酰化 | HDAC1/8→MHC-I/II↓、PD-L1调控异常 | 免疫抑制微环境维持、T细胞耗竭 | Entinostat+ipilimumab/nivolumab(NCT02453620) | I-II期临床 |
CD55+ MDSC扩增 | CD55-PYGL→糖酵解/STAT3激活 | MDSC免疫抑制功能维持、抗PD-1耐药 | Tomatidine(CD55-PYGL破坏剂)联合ICI | 临床前 |
上表汇总了乳腺癌免疫治疗耐药的八大核心机制及其对应的克服策略。从表中可以识别出三个关键趋势:第一,代谢-表观遗传-免疫三重交叉是耐药网络的核心特征——ISR通路同时调控代谢和免疫,EZH2/DNMT既影响表观遗传又调控免疫基因表达,提示联合治疗需要同时覆盖多个层面;第二,从临床前到临床转化的”死亡之谷”在免疫联合治疗中尤为突出——多数表观遗传和代谢靶向药物仍处于I-II期临床阶段,其安全性和最佳剂量方案在人体中尚未确立;第三,BRCA突变/HRD状态是乳腺癌中少数经过验证的ICI疗效预测标志物之一,PARP抑制剂与ICI的联合在BRCA突变人群中展现出最高的客观缓解率,为”合成致死+免疫”的精准策略提供了范例。
5.4 免疫系统与治疗策略的整合
5.4.1 “冷肿瘤”到”热肿瘤”转化策略
ER+乳腺癌(尤其低TILs亚群)本质上属于”冷肿瘤”,免疫治疗获益有限。将”冷肿瘤”转化为”热肿瘤”的策略正在从概念走向临床验证。Zhang等人的研究表明,低剂量decitabine可逆转免疫基因的表观遗传沉默,恢复ISGs表达并增加功能性TILs浸润。GCN2抑制剂(GCN2iB)通过解除氨基酸感知介导的T细胞功能抑制,恢复效应T细胞功能并与抗PD-1抗体协同。此外,支链氨基酸(Branched-Chain Amino Acids, BCAAs)补充通过提升循环NK/NKT细胞水平增强ICI疗效——在E0771乳腺癌模型中,BCAA补充联合ICI使肿瘤体积减少从单药ICI的44.1%提升至74.2%;在高度ICI耐药的4T1模型中,BCAA联合ICI实现了51%的肿瘤减少(单药ICI几乎无效)。这些转化策略的共性在于,它们并非直接杀伤肿瘤细胞,而是通过重塑TME的免疫状态恢复宿主抗肿瘤免疫,因此更适用于联合ICI的”增敏”场景而非单药治疗。
5.4.2 免疫治疗时空窗口
跨维度临床数据一致显示,ICI在乳腺癌中的获益高度依赖于治疗时机和肿瘤生物学特征。KEYNOTE-522(新辅助pembrolizumab+化疗)最终总生存期(Overall Survival, OS)分析显示5年OS率86.6% vs 81.7%(P=0.002),而ALEXANDRA/IMpassion030(术后辅助atezolizumab)则完全失败(侵袭性无病生存期HR 1.11,95% CI 0.87-1.42)。这一鲜明对比提示,新辅助阶段肿瘤抗原完整存在、可激活肿瘤特异性T细胞,是ICI获益的关键生物学窗口;辅助阶段肿瘤已切除、缺乏抗原刺激,可能大幅削弱免疫治疗的生物学基础。BELLINI试验进一步验证了TILs作为去化疗免疫治疗筛选标志物的可行性:对于TILs≥50%的早期TNBC患者,仅6周nivolumab+ipilimumab(无化疗)即可获得33%的病理完全缓解(Pathologic Complete Response, pCR)率和53%的主要病理缓解率。这些发现共同勾勒出一幅”免疫治疗时空窗口”图景:新辅助阶段优于辅助阶段、高TILs肿瘤优于低TILs肿瘤、TNBC优于ER+乳腺癌——免疫治疗的成功需要同时满足”时间窗口”和”肿瘤免疫状态”两个条件。
5.4.3 免疫-ADC联合
抗体药物偶联物(Antibody-Drug Conjugate, ADC)与ICI的联合正在乳腺癌中展现出协同潜力。ASCENT-04/KEYNOTE-D19 III期试验(2025年ASCO报告)证实,sacituzumab govitecan(SG)+pembrolizumab在PD-L1+ mTNBC一线治疗中显著优于化疗+pembrolizumab(中位无进展生存期11.2个月 vs 7.8个月,HR 0.65,P<0.001),成为首个ADC联合ICI在一线mTNBC中显示临床获益的III期试验。I-SPY2.2试验中,datopotamab deruxtecan+durvalumab在Immune+患者中的pCR率达65%,加用化疗后升至77%。NeoSTAR试验则探索了新辅助SG+pembrolizumab的去蒽环类策略,4周期pCR率32%,加用非蒽环类化疗后升至50%。ADC-ICI协同的可能机制包括:ADC杀伤释放肿瘤抗原增强免疫原性、ICI解除T细胞抑制恢复对ADC释放后抗原的应答,以及两者共同重塑TME。然而,3-4级不良事件发生率较高(ASCENT-04中71%)提示毒性管理是该联合策略的临床挑战。
5.4.4 肠道微生物组与免疫治疗
肠道微生物组通过多种机制调控乳腺癌免疫治疗响应。Ullern等人在Molecular Oncology发表的研究证实,肠道微生物组多样性是mTNBC化疗-免疫治疗获益的独立预后因素。AMTEC试验(NCT03801369)进一步发现,olaparib治疗后alpha多样性高的患者更可能从durvalumab联合治疗中获益。2025年发表于JCI Insight的研究揭示了微生物来源BCAAs通过提升循环NK/NKT细胞水平增强ICI疗效的分子机制——减重手术后的粪菌移植可将ICI获益转移至受体小鼠,BCAA被确认为关键效应分子。与ICI有利响应相关的菌群包括Faecalibacterium prausnitzii、Akkermansia muciniphila和短链脂肪酸(Short-Chain Fatty Acids, SCFAs)产生菌;而不利响应则与Prevotella copri和Enterobacteriaceae相关。这些发现提示,维持治疗期间肠道微生物组多样性(如避免不必要的抗生素使用)可能是一项简单可行的免疫治疗优化措施,但微生物组干预(粪菌移植、益生菌补充)的标准化方案和最佳时机远未确定。
5.4.5 精准免疫治疗生态系统
乳腺癌免疫治疗正在从单一的ICI应用向多维度精准生态系统演进。这一生态系统的核心要素包括:以TILs/PD-L1/TMB为代表的传统免疫标志物、以空间生态型和ISR签名为代表的新型标志物、以低剂量表观遗传药物和代谢调控剂为代表的”冷-热”转化工具、以ADC和CAR-T为代表的新型免疫药物,以及以肠道微生物组和个体化疫苗为代表的系统性免疫调节策略。KEYNOTE-522确立了早期TNBC新辅助免疫治疗的标准地位,NeoPACT试验验证了无蒽环类新辅助免疫化疗方案的可行性(pCR率58%,3年无事件生存率86%),BELLINI试验为去化疗策略提供了概念验证,而TNBC-MERIT试验则首次证明个体化mRNA新抗原疫苗在乳腺癌辅助治疗中的可行性和持久T细胞免疫原性(14/15例完成治疗,平均生产周转69天)。这些试验的组合描绘了一个多层次、个体化的精准免疫治疗图景:初诊时通过TILs、分子分型和空间生态型评估免疫状态;治疗中选择新辅助免疫治疗(±化疗、±ADC);治疗中监测免疫编辑状态和ctDNA动态;耐药时通过表观遗传/代谢/微生物组干预恢复免疫敏感性。需要指出的是,这一”生态系统”的多数组件仍处于早期验证阶段,其临床落地的标准化路径和成本效益尚待确立。
6. 总结与展望
6.1 关键进展总结
6.1.1 精准治疗闭环:从分子分型到液体活检动态管理的全周期精准模式
乳腺癌精准治疗正在完成从”静态分型”到”动态闭环”的范式跃迁。FUTURE-SUPER试验验证的TNBC分子分型指导治疗与SERENA-6试验确立的ctDNA动态监测指导治疗切换构成了这一闭环的两端——前者解决初诊时的治疗方案选择问题,后者解决治疗中的耐药监测与方案调整问题。inavolisib三联方案在PIK3CA突变患者中的成功进一步将分子分型与靶向治疗紧密耦合。这三个标志性试验的交汇意味着,乳腺癌治疗正逐步形成”初诊全基因组/转录组分型→一线精准方案→ctDNA动态监测→分子进展早期干预→耐药后再分型”的全周期精准管理模式。然而,这一闭环的大规模临床落地仍面临检测标准化、数据整合平台和成本效益等现实挑战,特别是在资源有限环境中,基于四种IHC标志物(AR/CD8/FOXC1/DCLK1)的简化分型方案可能是更务实的过渡策略。
6.1.2 免疫治疗突破:从TNBC扩展到ER+,从辅助到新辅助的范式转变
免疫检查点抑制剂(Immune Checkpoint Inhibitor, ICI)在乳腺癌中的发展历程揭示了一个深刻的生物学规律:免疫治疗的成功需要同时满足”时间窗口”和”肿瘤免疫状态”两个条件。新辅助阶段肿瘤抗原完整存在,可激活特异性T细胞,这是KEYNOTE-522取得5年OS获益和CamRelief实现pCR率56.8%的生物学基础;而辅助阶段肿瘤已切除、缺乏抗原刺激,则解释了ALEXANDRA试验的完全失败。ER+乳腺癌中CheckMate 7FL和KEYNOTE-756的阳性结果虽然将ICI的边界从TNBC扩展至HR+领域,但pCR绝对提升仅约10%,且主要集中于ER低表达(1%—9%)和PD-L1高表达亚组,提示ER+亚型本质上属于免疫”冷肿瘤”,需要”冷→热”转化策略才能充分释放免疫治疗的潜力。BELLINI试验中TILs≥50%患者仅接受6周免疫治疗即获33%的pCR率,为去化疗免疫治疗提供了概念验证,也为精准筛选免疫治疗获益人群确立了可行路径。
6.1.3 ADC治疗变革:覆盖HER2-low/ultralow至一线治疗的全谱系应用
ADC药物正在重塑乳腺癌全亚型的治疗版图。DESTINY-Breast09将HER2+晚期乳腺癌一线PFS标准提升至40.7个月,DESTINY-Breast06将T-DXd的适应人群扩展至HER2-ultralow,两者结合使T-DXd覆盖了约85%的乳腺癌患者。ASCENT-04开创的ADC+ICI联合模式将PD-L1阳性mTNBC一线PFS提升至11.2个月,而SKB264作为中国原研ADC的成功上市则标志着全球ADC竞争格局已从欧美垄断转向多极化。值得关注的是,三种已获批ADC均采用拓扑异构酶I(Topoisomerase I, TOP1)抑制剂载荷,TOP1突变介导的交叉耐药已成为序贯治疗中的现实挑战,开发非TOP1载荷ADC和基于ctDNA的耐药监测将是优化序贯策略的关键方向。ADC药物虽本质为广谱细胞毒性载荷递送系统,但HER2/TROP2表达水平与疗效的关联性日益明确,提示ADC正在从”一刀切”向标志物驱动的精准化演进。
6.2 未来方向
6.2.1 AI驱动的多组学整合
人工智能(Artificial Intelligence, AI)在乳腺癌中的应用正从单一影像辅助向多组学整合决策演进。MASAI试验已证实AI在筛查中的临床价值,而AI整合基因组学、转录组学、蛋白质组学和ctDNA动态数据的”虚拟肿瘤委员会”正在成为研究前沿。FUSCC-TNBC分型和SNF 4亚型的建立依赖于多组学整合算法,未来AI驱动的实时整合可能实现治疗方案的动态优化——基于治疗中ctDNA的突变谱变化、影像组学特征演变和临床参数联合建模,预测最佳治疗切换时机。然而,多组学AI模型的临床验证需要大规模前瞻性队列支持,且数据隐私、算法可解释性和监管框架等议题尚待解决。
6.2.2 “去化疗”策略
“去化疗”策略正从概念走向循证实践。BELLINI试验中TILs≥50%的TNBC患者仅接受免疫治疗即可获得33%的pCR率,ADAPT系列试验基于Ki67反应的降阶梯策略正在验证中危HR+患者免除化疗的可行性,NeoPACT试验证实无蒽环类新辅助免疫化疗方案可达到58%的pCR率。这些试验共同勾勒出去化疗的可行性路径:通过精准生物标志物(TILs≥50%、低Oncotype DX评分、Ki67反应良好、影像组学低危特征)筛选低化疗需求人群。初步估算,约20%—30%的TNBC患者和相当比例的HR+早期患者可能具备去化疗条件,但长期随访数据尚待成熟,以确认去化疗策略是否不劣于标准方案的无病生存期和总生存期。
6.2.3 细胞治疗与疫苗
个体化新抗原疫苗和TIL疗法代表了乳腺癌免疫治疗的最前沿方向。TNBC-MERIT试验首次证明个体化mRNA新抗原疫苗在乳腺癌辅助治疗中的可行性和持久T细胞免疫原性,为疫苗联合ICI的协同策略奠定了基础。TIL疗法在黑色素瘤中的成功正逐步向乳腺癌延伸,联合ICI的初步研究显示1例转移性乳腺癌患者获得完全缓解,但TIL扩增效率、制造周期和成本仍是规模化应用的主要障碍。CAR-T疗法在实体瘤中的挑战更为严峻——抗原异质性、脱靶毒性和TME免疫抑制构成三重壁垒,目前大多数CAR-T乳腺癌试验仍处于I期阶段。细胞治疗和疫苗的临床转化可能需要5—10年的循证积累。
6.2.4 全球公平性
乳腺癌精准治疗的全球可及性面临严峻挑战。低人类发展指数(Human Development Index, HDI)国家与高HDI国家之间存在4—5倍的年龄标准化发病率差异和更为悬殊的死亡率差异,ctDNA检测、多基因表达谱分析和ADC药物等精准治疗工具在中低收入国家的可及性极为有限。亚洲地区占全球乳腺癌新发病例的42.9%,中国5年生存率虽持续改善但与欧美仍有差距。FUSCC-TNBC分型系统和CamRelief试验代表的中国原创研究为全球贡献了基于亚洲人群的治疗证据,但精准治疗基础设施的区域间不平衡仍是全球乳腺癌防控的核心挑战。未来需要在简化检测平台(如基于IHC的分型替代基因组学分型)、仿制药和生物类似药普及、以及国际多中心试验纳入中低收入国家等方面进行系统性投入。
6.3 临床转化建议
6.3.1 基于证据的治疗决策框架
当前乳腺癌治疗决策面临的核心挑战是循证证据的快速增长与临床实践转化之间的时滞。基于本报告综述的数据,建议构建分层决策框架:对于早期TNBC,新辅助免疫治疗+化疗已成为标准(KEYNOTE-522 5年OS获益明确),TILs≥50%的患者可考虑去化疗免疫治疗(BELLINI模式),术后辅助免疫治疗的证据为阴性(ALEXANDRA),不推荐常规使用。对于HER2+晚期乳腺癌,T-DXd+帕妥珠单抗已取代THP成为一线标准(DESTINY-Breast09)。对于HR+/HER2-晚期乳腺癌,一线CDK4/6抑制剂+内分泌治疗地位稳固,ctDNA监测ESR1突变可指导早期切换至口服选择性雌激素受体降解剂(Selective Estrogen Receptor Degrader, SERD)(SERENA-6模式),PIK3CA突变患者可考虑inavolisib三联方案。对于mTNBC,PD-L1阳性患者首选SG+pembrolizumab(ASCENT-04),PD-L1阴性患者可考虑SG或化疗。HER2-low/ultralow患者应接受T-DXd而非化疗(DESTINY-Breast06)。这一框架需结合分子标志物检测的可及性进行本土化调整。
6.3.2 多学科协作与个体化治疗路径
乳腺癌精准治疗的复杂性已超越单一学科的决策能力。分子分型(FUSCC/SNF)需要病理科和分子诊断平台的支持,ctDNA动态监测需要检验科和生物信息学的协作,ADC和免疫治疗的不良事件管理需要内科、呼吸科和风湿免疫科的联合参与,而去化疗决策则需要影像科、外科和肿瘤内科的集体评估。建议建立以患者为中心的”精准乳腺肿瘤委员会”模式,整合分子病理、影像组学、液体活检、临床药学和患者报告结局等多维度信息,制定个体化治疗路径。治疗路径应遵循”标志物驱动、动态调整、降阶梯优先”的原则:初诊时进行全面分子分型,治疗中每2—3个月进行ctDNA监测(在资源允许条件下),耐药时进行再活检以捕获亚克隆演化,同时根据TILs、Ki67反应和影像组学特征评估降阶梯/去化疗的可行性。这种多学科整合模式不仅是技术层面的要求,更是实现乳腺癌全周期精准管理的组织保障。