


从非美国对照药到PK研究豁免:Revision 4如何重塑$2320亿市场的竞争规则

2026年3月9日,FDA再补一刀
FDA发布了《生物类似药开发与BPCI法案问答(第四版修订草案)》——这份以Q&A形式呈现的行业指南,是FDA在半年内连续第三次降低生物类似药开发门槛。
2025年10月,FDA砍掉了临床效能比较研究(CES, comparative efficacy study)的强制要求。2024年6月,FDA提出取消互换性切换研究。这次Revision 4更进一步:非美国对照药(non-U.S.-licensed comparator product)的临床数据可以在科学论证充分时直接用于美国申报,PK研究中也不再强制要求与美国参照药(U.S.-licensed reference product)直接比较。
三刀叠加的效果:FDA估计,仅PK研究简化一项,就能为每个生物类似药开发项目节省约$2000万,即约50%的PK研究成本。加上CES的取消,总开发费用可缩减$1.5亿以上,周期缩短2-4年。
问题是:即便门槛降了,2025-2034年到期的118个生物药中,90%仍然没有类似药在研。FDA是在跟一个$2320亿的结构性空白赛跑。
一、Revision 4改了什么?三条Q&A的精准手术

Q&A I.8:非美国对照药的数据准入
这是本次修订的核心。过去,申请人如果用非美国许可的对照药做了临床PK研究,FDA要求额外做一个"三方桥接试验"(three-way PK study):把拟议生物类似药、美国参照药、非美国对照药放在一起比。成本高、周期长、对全球多中心试验极不友好。
Revision 4拆成两部分。Part (a)明确:如果申请人能提供充分科学论证,证明非美国对照药与美国参照药之间的关联性,那么用非美国对照药做的PK或其他临床数据可以直接支持相似性证明——不需要三方桥接。甚至取消了"至少一项PK研究必须直接与美国参照药比较"的旧要求。
Part (b)聚焦分析比对。FDA希望看到非美国对照药与美国参照药之间的比较分析数据(comparative analytical data),作为科学论证的基础。FDA还专门就"分析比对的实用性"征求公众意见——这个信号说明,FDA内部对这条路径的具体操作标准仍在校准。
Q&A I.10:临床研究留样保存
修订后的Q&A I.10对比较临床研究中使用的产品留样(reserve samples)提出了更新的保存期限和数量要求。目的是确保研究的可追溯性,同时与国际标准对齐,降低全球多中心试验的合规负担。
Q&A I.19:强度与活性成分含量一致性
FDA进一步明确了"相同强度"(same strength)的定义。对注射用产品,"强度"指的是每个容器中药物的总含量(total drug content),而非浓度。这给了申请人更大的灵活度——只要总递送剂量一致,预充式注射器或自动注射器的设计可以有差异。
二、IND申请与标签规范:非美国对照药的实操路径
在美国境内开展比较临床试验时,非美国许可的对照药在美国法律下属于研究性新药。Revision 4明确了几个操作要点:
问题 | FDA立场 |
非美国对照药是否需要IND? | 是。在美国使用的非美国对照药属于investigational drug,需要IND申请 |
能否合并IND申请? | 可以。申请人可将拟议生物类似药和对照药纳入同一个IND |
CMC数据不完整怎么办? | 可申请豁免。对照药制造商的CMC信息通常难以获得,FDA允许在条件不具备时请求信息豁免 |
标签怎么标? | 对照药须按IND规定标注,不得以商业产品标签进入临床试验 |
这套框架的实际意义:一家韩国或印度的生物类似药企业,可以先在本国用当地参照药完成PK和临床研究,再把数据带到美国申报——前提是提供充分的分析比对,证明本国参照药与美国参照药的关联性。不需要在美国重做一遍PK。
对比过去的路径,这省掉了一次完整的临床PK研究成本——按FDA估算约$2000万和至少一年时间。对开发多个生物类似药品种的企业来说,这个政策的杠杆效应是指数级的。
三、政策背景:三刀连环与2015指南的正式退场
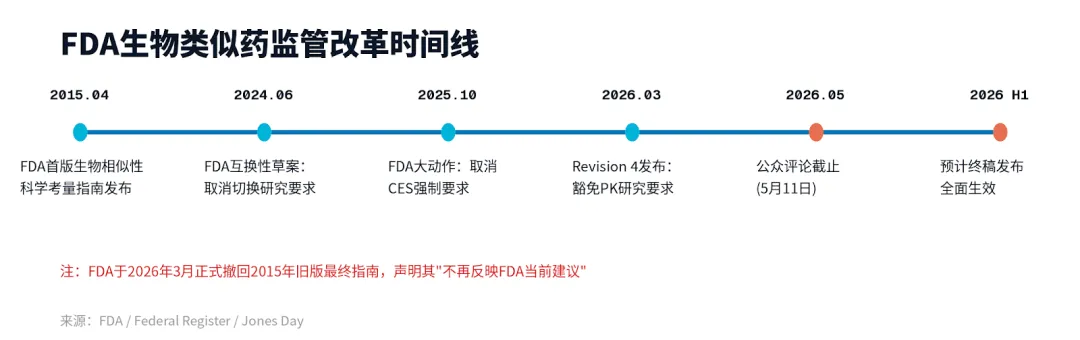
Revision 4不是孤立事件,是FDA过去18个月连续政策动作的第三步。
时间 | 动作 | 影响 |
2024年6月 | 互换性草案:取消切换研究 | 所有获批生物类似药默认可互换,药剂师可直接替代 |
2025年10月 | CES豁免草案:取消临床效能比较 | 省掉$1-1.5亿开发成本和1-3年周期 |
2026年3月 | Revision 4:PK研究简化 + 非美国对照药准入 | 再省$2000万/项目,全球开发数据可直接利用 |
同日 | 正式撤回2015年旧版指南 | "不再反映FDA当前建议"——旧时代彻底结束 |
FDA局长Marty Makary的原话很直接:"用常识来推动更精确的分析技术路径。"HHS部长把旧体系形容为"天价成本、无尽红线"。
政治背景也不能忽略。特朗普政府在降药价议程上需要可见的成果。生物类似药是交集——它同时满足"降价""去监管""促竞争"三个政策偏好。FDA在半年内连发三份草案,跟这个政治节奏高度同步。
四、$2320亿的结构性空白:降门槛够吗?
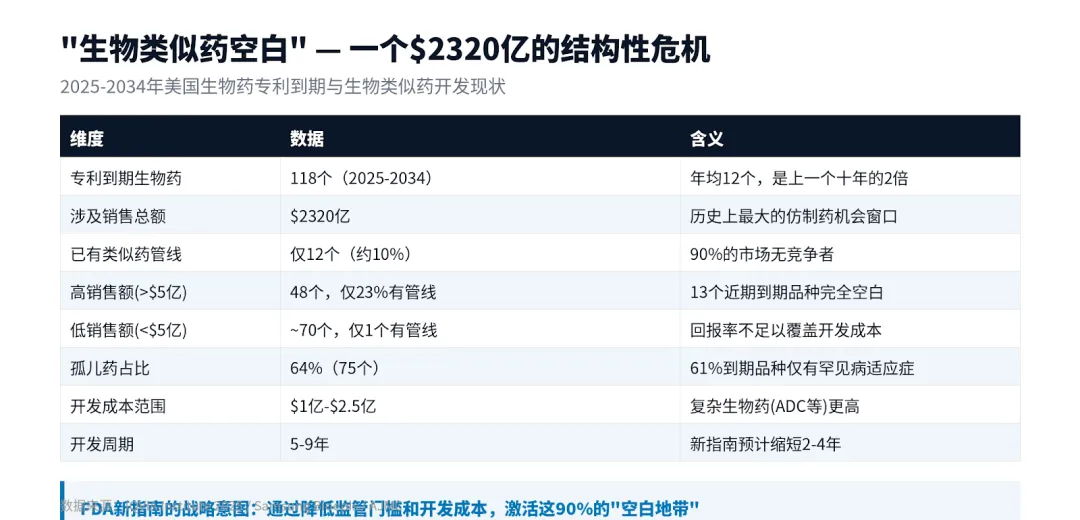
IQVIA的数据很扎眼:2025-2034年间,118个生物药将失去专利保护,涉及$2320亿美国销售额。但截至2024年中,只有12个有生物类似药在研——覆盖率仅10%。
为什么门槛降了,开发者还是不来?几个结构性原因:
回报率不够。低销售额生物药(年销售低于$5亿)占到期品种的60%,但几乎没有类似药管线。$1-2.5亿的开发成本对小市场品种来说,账算不过来。
复杂度太高。下一代生物药——抗体偶联药物(ADC)、双特异性抗体、细胞疗法——对类似药开发者的制造能力和CMC控制提出了远超传统单抗的要求。2025-2034年有16个复杂生物药将到期,目前零个有类似药在研。
孤儿药占比过高。64%的到期品种涉及孤儿适应症。患者群体小,市场天花板低,开发动力天然不足。
IRA带来的不确定性。《通胀削减法案》下的Medicare药价谈判改变了生物药的收入预期。如果参照药已被谈判压价,类似药的价差优势进一步缩窄。
FDA的监管简化能解决的是"时间和成本"这一层障碍。但市场回报率、制造复杂度和支付环境这些结构性问题,不是指南草案能修复的。
五、对中国企业的启示
Revision 4对中国生物类似药企业是一个实质性利好。
过去,中国企业在中国或其他国家用当地参照药完成的PK和临床研究,到了美国申报时基本需要重做。三方桥接试验的要求意味着全球开发数据不能直接互认。
现在路径打开了:如果能提供非美国对照药与美国参照药之间充分的分析比对数据,中国或欧洲的临床数据有可能直接被FDA接受。这降低了中国企业赴美申报的边际成本——尤其对已经在国内完成了高质量PK研究的品种。
关键前提是"科学论证的充分性"。FDA在征求意见中专门提问:分析比对在多大程度上能替代临床桥接?这说明具体标准还没定死。先行者可以通过Pre-BLA会议跟FDA确认个案要求,占据信息优势。
结语
FDA用三份草案在18个月内拆掉了生物类似药开发路径上三道最贵的关卡。但$2320亿的空白地带提醒我们:监管只是门槛之一,市场动力才是真正的引擎。
Revision 4的公众评论期到2026年5月11日。如果按FDA的节奏,上半年内可能出终稿。届时,2015年指南时代遗留的最后一批规则也将被正式替换。
对生物类似药开发者来说,现在是审视全球数据资产、重新规划美国申报策略的窗口期。对临床研究者来说,留样保存和IND程序的更新需要嵌入方案设计。对政策观察者来说,真正的看点不在指南本身——而在指南能不能在这十年里把那个90%的空白填上哪怕一小部分。
免责声明:本文基于FDA公开文件、Federal Register公告及行业分析整理,仅供专业科普与参考,不构成任何医疗或投资建议。监管政策最终以FDA终稿为准。
主要引用来源:FDA Federal Register 2026-04664 / FDA.gov指南文件 / IQVIA Institute 2025 / Big Molecule Watch / The FDA Law Blog / Jones Day / Latham & Watkins / AJMC / Fierce Pharma / ASCO Post
欢迎添加博主微信交流
