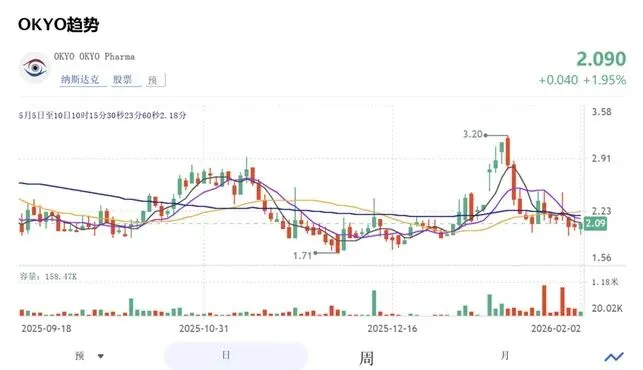
1.1 NCP的定义、病理生理学与独特性


外周敏化: 角膜神经末梢因损伤(如屈光手术、病毒感染、化学灼伤)或长期炎症(如严重干眼症)而变得过度兴奋,导致对正常刺激(如眨眼、风吹)的反应增强,甚至产生自发性疼痛。
中枢敏化: 持续的外周疼痛信号输入,导致脊髓三叉神经核和更高级的中枢神经系统(如丘脑、皮层)发生可塑性改变。这使得中枢神经系统对疼痛信号的“放大”效应增强,并降低了疼痛阈值,导致疼痛的慢性化和顽固化,即使外周刺激已经消除,疼痛感依然存在 (根据背景信息)。
1.2 临床困境:诊断挑战与巨大的未满足需求


局部疗法: 大剂量无防腐剂人工泪液、自体血清滴眼液、局部应用的皮质类固醇或免疫抑制剂(如环孢素、他克莫司)等,这些疗法主要针对眼表炎症,对神经病理性疼痛本身效果有限 (根据背景信息)。
全身性药物: 抗惊厥药(如加巴喷丁、普瑞巴林)、三环类抗抑郁药(如阿米替林、去甲替林)等。这些药物虽是治疗其他神经痛的常用药,但用于NCP时,常因全身性副作用(如嗜睡、头晕、口干)而导致患者耐受性差,且疗效不一。
介入性治疗: 巩膜接触镜、肉毒杆菌毒素注射、神经调节或阻滞等,这些方法或为侵入性,或证据等级不高,难以作为一线标准化治疗方案。
2.1 药物基本信息与创新作用机制(MoA)


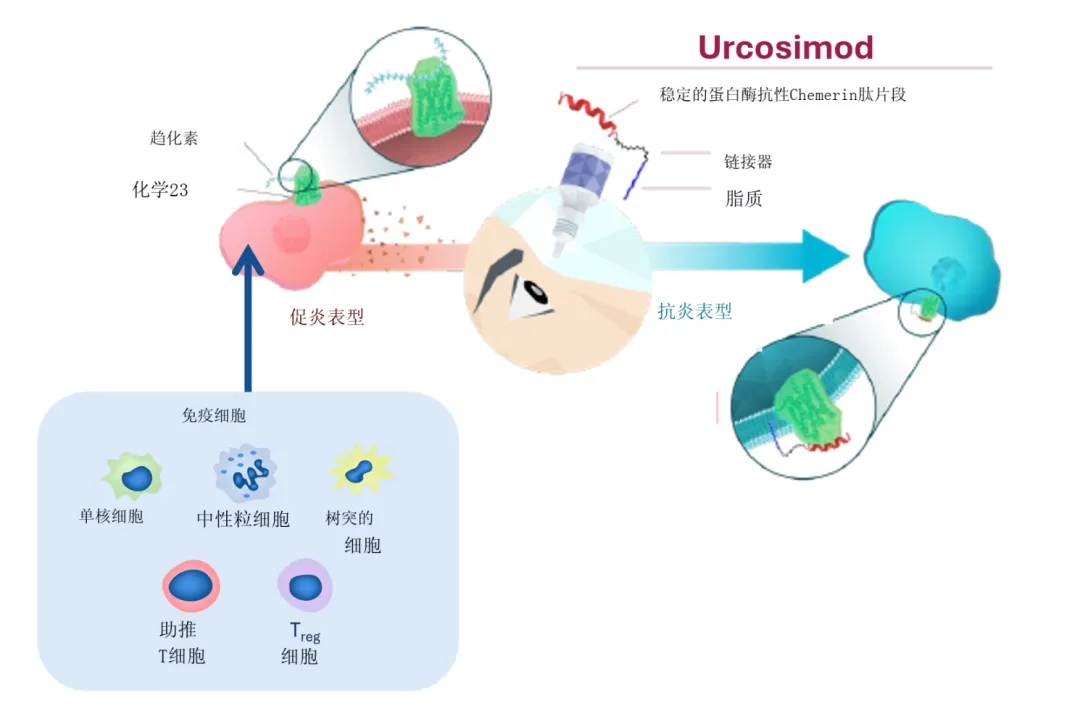
药物名称: Ucosimod (乌司莫德,研发代码:OK-101)
药物类别: ChemR23(趋化素受体23)肽激动剂 (根据背景信息)
分子设计: Ucosimod是一种经过精心设计的 脂质结合肽(Lipid-tethered peptide) 。根据OKYO Pharma披露的信息,其分子结构可能包含三个部分:一个与内源性配体Chemerin(趋化素)活性序列相似的 肽片段 ,负责激动ChemR23受体;一个 脂质锚(Anchored Lipid) ,用于将药物分子“钉”在眼表细胞膜上,从而显著延长其在泪膜和角膜组织的驻留时间;以及一个连接两者的 连接子(Linker) 。这种设计旨在克服传统肽类药物在眼部快速清除的难题,实现低频给药和高效靶向。
作用靶点与机制: Ucosimod的核心靶点是 G蛋白偶联受体ChemR23 。该受体在免疫细胞(如巨噬细胞、树突状细胞)以及神经元和神经胶质细胞中均有表达。
抗炎作用: Chemerin/ChemR23信号通路是一个关键的炎症“消退”开关。在炎症初期,它可能促进免疫细胞募集;但在炎症后期,激活ChemR23能够启动“促消退(Pro-resolving)”程序,抑制促炎细胞因子的产生,促进抗炎介质的释放,从而主动地终止炎症反应,而非被动抑制 (根据背景信息)。在NCP的背景下,控制角膜微环境的慢性神经炎症,被认为是减轻角膜神经末梢外周敏化的关键。
直接镇痛作用: 更重要的是,ChemR23受体同样在背根神经节(DRG)等感觉神经元上表达。临床前研究表明,激动ChemR23可能直接抑制伤害感受性神经元的兴奋性,从而产生镇痛效应。这意味着Ucosimod可能具备“抗炎”和“镇痛”的双重作用,这对于治疗兼具炎症和神经病理成分的NCP而言,是一个极具吸引力的特性。
2.2 临床前及早期临床研究概览


临床前研究: 在多种眼部疼痛和炎症的动物模型中,Ucosimod展示了良好的抗炎和镇痛活性 (根据背景信息)。这些研究为该药物进入人体试验提供了理论基础。
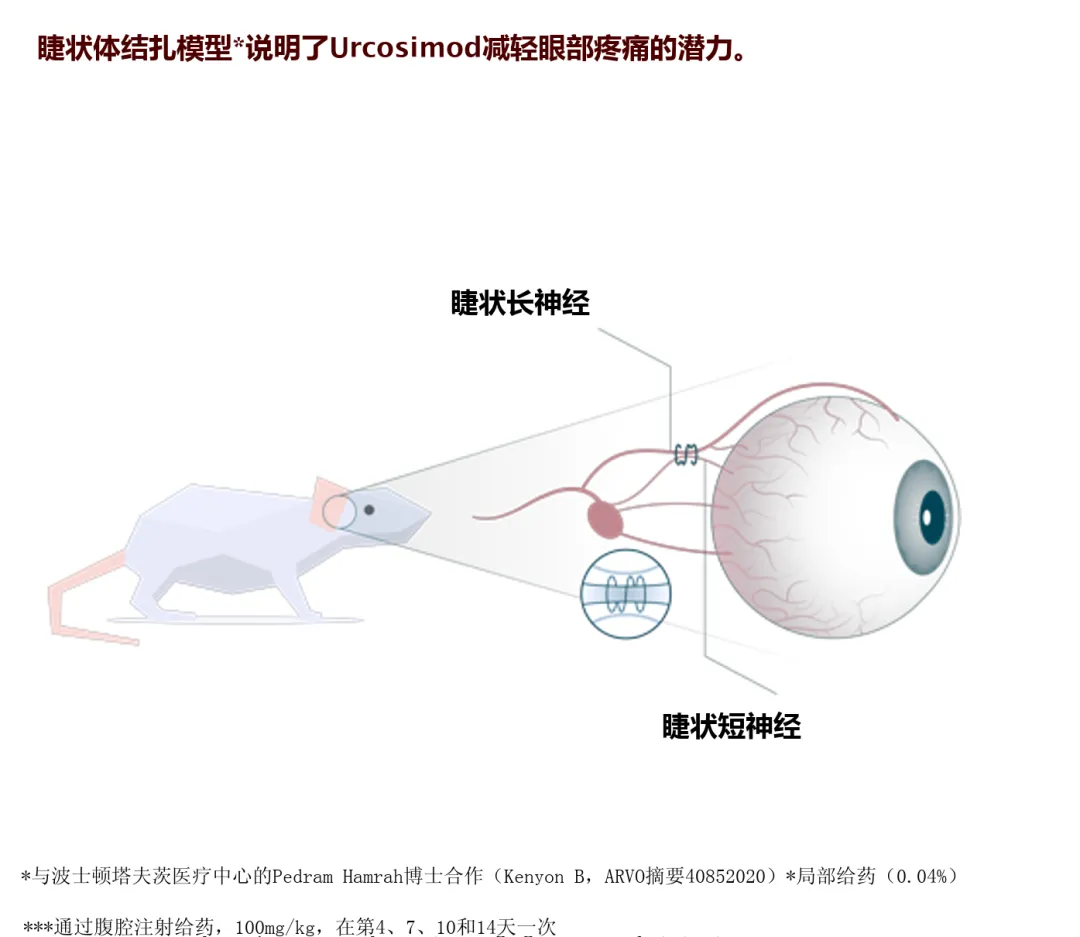
干眼症 (DED)2 期试验: 公司报告了一项针对240名DED患者的大型2期试验取得了积极结果,达到了其主要和次要终点 (根据背景信息)。尽管DED与NCP不完全相同,但两者常有重叠,该试验的成功为Ucosimod在眼表的安全性和抗炎/镇痛潜力提供了初步的人体证据。
NCP 2期试验: 公司同样报告了一项针对18名NCP患者的小型、随机、双盲的2期试验获得了积极结果 (根据背景信息)。虽然样本量很小,但这一探索性研究的结果很可能是促使公司与FDA召开C类会议,并设计更大规模试验的直接原因。
3.1 C类会议在FDA药物审评中的战略价值


3.2 主要终点的确立:VAS评分降低≥2分的临床意义


什么是VAS? 视觉模拟量表 是一种广泛用于测量主观感受(如疼痛、焦虑)的工具。它通常是一条10厘米(或100毫米)的直线,一端标为“无疼痛”,另一端标为“无法忍受的最剧烈疼痛”。患者在线上标记出能代表其当前疼痛程度的位置,通过测量标记点到起点的距离,将主观感受量化为0-10(或0-100)的数字。
为何“≥2分”具有里程碑意义?
3.3 试验设计的认可:2b/3期适应性设计的战略价值


2b/3期适应性设计 :这是一种高效的临床试验策略。它通常在一个试验方案下,无缝衔接地包含两个阶段。
2b阶段(剂量探索): 试验前期,多个剂量的Ucosimod组与安慰剂组进行比较,主要目的是确定哪个剂量在疗效和安全性上表现最佳(即“剂量-效应关系”)。
3期阶段(关键性验证): 在中期分析后,可能会根据预设规则,将表现不佳的剂量组剔除,然后继续招募患者进入已确定的最佳剂量组和安慰剂组,以足够的样本量来最终验证疗效。
节省时间与成本: 相比于先完成一个独立的2期试验,再设计和启动一个全新的3期试验,适应性设计将两个阶段融合,大大缩短了研发周期。
提高成功率: 通过在试验中动态调整,可以选择最优剂量进入关键性验证阶段,避免了因2期剂量选择不当而导致3期失败的风险。
对于NCP这类罕见或难招募患者的疾病尤为适用 ,因为它能更有效地利用每一位入组的宝贵患者资源。
3.4 次要终点与统计学考量


次要终点:眼部疼痛评估调查(OPAS) :FDA支持使用OPAS作为次要终点,这一点同样意义重大 (根据背景信息)。OPAS是一个经过验证的、专门用于评估眼部疼痛对患者 生活质量和功能(如阅读、驾驶、使用电脑)影响的问卷。将其纳入,意味着FDA不仅关心患者疼痛分数的降低,更关心这种降低能否转化为实际生活功能的改善 。这使得对Ucosimod疗效的评估更加立体和全面。
统计学指导: FDA提供了旨在增强分析稳健性的统计学指导,并指出,若统计分析计划(SAP)在揭盲前最终确定且结果有说服力,单次关键性试验的数据就可能支持有效性结论 (根据背景信息)。这暗示了如果试验结果足够“干净”和“强大”,Ucosimod或许有机会仅凭这一个2b/3期试验的数据就申请上市,这无疑是一个巨大的潜在利好。
3.5 CMC与快速通道资格的加持


CMC(化学、制造和控制): FDA对Ucosimod的CMC策略未提出任何重大关切 (根据背景信息)。这意味着FDA对药物的生产工艺、质量控制、稳定性等方面初步满意。CMC是新药研发中常被忽视但至关重要的一环,许多有前途的药物都因CMC问题而受阻。在此阶段获得FDA的认可,表明Ucosimod的“产品化”进程相对顺利。
快速通道资格: Ucosimod此前已获得FDA授予的快速通道资格。这一资格授予旨在促进治疗严重疾病且具有潜力满足未满足医疗需求的药物的开发和加速审查。获得该资格意味着OKYO Pharma可以与FDA进行更频繁的沟通,并有机会获得 加速批准和优先审评 的资格,从而可能比常规流程更早上市。
4.1 对NCP药物研发的范式重塑


从“无路可走”到“有路可循”: 在此之前,任何想开发NCP药物的公司都面临一个根本性问题:“我该如何向监管机构证明我的药有效?”现在,FDA通过Ucosimod的案例,清晰地勾勒出了一条路径: 以患者报告的疼痛VAS评分为核心主要终点,以功能性量表OPAS为重要支持,采用严谨的随机对照试验设计。
靶点多样化的可能性: Ucosimod靶向ChemR23,验证了“抗炎+镇痛”双重机制的可行性。这会激励其他研发者探索更多与神经炎症和疼痛信号通路相关的创新靶点,如TRP通道、P2X受体、S1P受体等。
眼科与其他疼痛领域的交叉融合: FDA将广泛应用于全身性疼痛研究的终点(VAS评分≥2分)和理念引入NCP领域,预示着未来眼科疼痛药物的研发将更多地借鉴和融合神经科学与疼痛医学的成熟经验。
4.2 “以患者为中心”理念在眼科研发中的深化


PROs的崛起: 患者报告结局(PROs)正从次要或探索性终点,上升为可以支持药物批准的核心终点。对于干眼症、过敏性结膜炎等以主观症状为主要困扰的疾病,这一趋势将更加明显。
对临床实践的意义: 当药物的疗效评估与患者的感受直接挂钩时,获批的药物将更有可能在真实世界中为患者带来切实可感的获益,从而提高患者的治疗依从性和满意度。
4.3 研发策略的启示:早期、主动、高质量地与监管机构沟通


沟通前置: 不要等到所有研究都做完,才去面对监管机构的“大考”。在关键决策点(如启动关键性试验前),主动发起沟通,可以避免走弯路,节省巨额的研发成本和宝贵的时间。
准备充分: 向监管机构展示的数据和计划必须是高质量的。OKYO Pharma能够获得FDA的认可,前提是其拥有扎实的临床前数据、初步的临床信号以及一个经过深思熟虑、符合科学和统计学原理的后期开发计划。
问题导向: 与监管机构的沟通应该是具体的、有针对性的。例如,“我们计划用VAS作为主要终点,阈值设为2分,您是否同意其临床意义?”这种清晰的问题,更容易获得明确的、可操作的反馈。
5.1 中国NCP研究与治疗现状评估


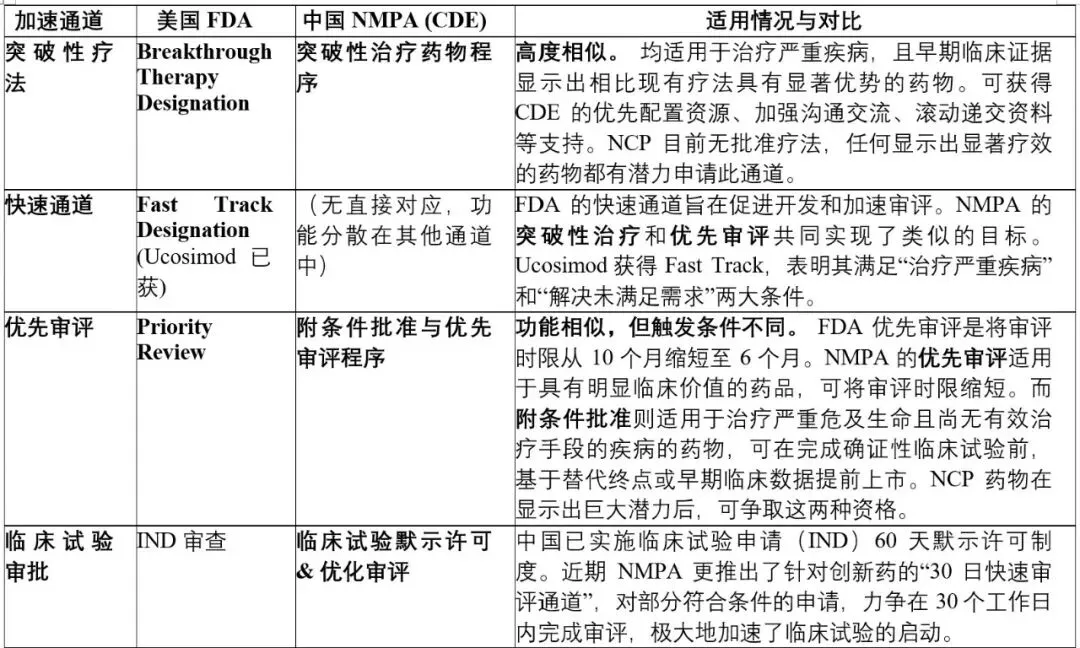
5.2 NMPA监管框架下的创新药加速路径


5.3 对国内眼科企业的战略建议


跳出“Me-too”思维: 与其在干眼症等竞争激烈的“红海”市场内卷,不如将目光投向NCP、葡萄膜炎、甲状腺相关眼病(TED)等存在巨大治疗空白的领域。这些领域虽然研发难度大,但一旦成功,临床价值和市场回报也更高。
深度挖掘病理机制: Ucosimod的成功源于对ChemR23通路在炎症消退和疼痛中作用的深刻理解。国内企业应加强基础研究投入,寻找和验证新的药物靶点,开发具有全球专利潜力的“First-in-class”或“Best-in-class”产品。
引入并验证PROs: 在临床试验设计初期,就应考虑引入国际通用的、经过验证的患者报告结局量表(如VAS, OPAS, OSDI等)。如果国内缺乏验证版本,应尽早开展文化调适和信效度验证工作,并就其作为关键终点的可行性与CDE进行沟通。
借鉴Ucosimod的终点标准: 在设计NCP或类似眼科疼痛疾病的临床试验时,可主动参考“VAS评分降低≥2分”这一已被FDA认可的临床意义阈值,作为自己的疗效目标,这将大大增加方案获得监管机构认可的可能性。
拥抱创新试验设计: 积极探索适应性设计、主协议(Master Protocol)等高效临床试验模式,尤其是在患者招募困难的罕见病或细分领域,以提升研发效率。
学习“C类会议”精神: 充分利用NMPA药品审评中心(CDE)提供的沟通会议机制。在项目关键节点,如Pre-IND阶段、关键性临床试验启动前,主动申请与CDE进行II类或III类沟通交流会议,就试验设计、终点选择等核心问题与审评员达成共识。
准备高质量的沟通材料: 与监管沟通的成功与否,取决于企业是否能提供逻辑严密、数据详实、问题明确的申报资料。这要求企业内部必须建立一支既懂研发又懂法规的专业团队。
关注创新递送技术: Ucosimod的脂质锚定技术是其成功的关键之一。国内企业应关注并布局眼科长效递送技术,如纳米胶束、原位凝胶、缓释植入剂等,以提升药物的生物利用度和患者依从性。
早期规划全球开发: 对于真正创新的眼科药物,应在立项之初就具备全球视野。考虑中美双报,利用ICH指导原则统一技术要求,争取在获得NMPA批准的同时,也能向FDA递交申请。虽然可能需要进行 桥接研究来评估种族差异 ,但全球同步开发的长远收益是巨大的 (根据背景信息中的桥接研究要求,虽然具体细节缺失,但其为国际化开发的必经之路)。
6.1 Ucosimod自身面临的挑战


临床试验结果的不确定性: FDA的积极反馈不等于最终批准。拟议的2b/3期试验尚未开始招募,其结果仍是未知的。疼痛类临床试验,尤其是NCP,因其强烈的安慰剂效应和患者异质性,失败率相对较高。
长期安全性未知: 局部长期使用一种ChemR23激动剂的潜在安全性风险(如对眼表免疫稳态、角膜愈合的影响)需要在更大规模、更长周期的临床试验中得到验证。
数据透明度问题: 如前所述,目前所有积极信息均来自公司单方面披露,缺乏同行评议的公开数据,这为客观评估带来了困难。
6.2 NCP研究领域的普遍难题


患者异质性: NCP的病因多样,不同患者的疼痛机制可能存在差异(例如,外周敏化为主 vs. 中枢敏化为主)。未来可能需要根据生物标志物对患者进行分层,以实现精准治疗。
客观生物标志物的缺失: 目前仍缺乏能够客观、可靠地反映NCP严重程度和治疗反应的生物标志物,这给新药研发的疗效评估带来挑战。
安慰剂效应的管理: 在疼痛临床试验中,安慰剂组的患者也常常报告显著的疼痛缓解。如何在试验设计和统计分析中有效地区分药物的真实疗效与安慰剂效应,是一个永恒的难题。
6.3 未来展望


更多靶向药物的出现: 针对不同疼痛通路的药物将被开发出来,为患者提供更多选择。
联合治疗方案的探索: 可能会出现将靶向神经的药物与抗炎或促进神经再生的药物联合使用的治疗新策略。
精准医疗的实现: 基于基因组学、蛋白质组学或影像学(如高分辨率共聚焦显微镜)的生物标志物将被发现,用于指导个体化的治疗选择。