





扫码进群学习交流
引言
牙周炎是一种高患病率、与生物膜相关的疾病,也是老年人群牙齿丧失的主要原因(Frencken 等人,2017)。在一项大型多中心研究(ABPARO)中,研究者探讨了阿莫西林(500 毫克)和甲硝唑(400 毫克)辅助牙周治疗的统计学显著获益,并发表了临床结果(Harks 等人,2015)。我们最近对来自 ABPARO 研究的非吸烟牙周炎患者进行了微生物亚组分析(Hagenfeld 等人,2018)。我们发现,在牙周治疗 2 个月后,所使用的抗生素导致了微生物组的转变,其特征是龈下生物膜中牙周病原体数量减少,共生细菌增加。吸烟是破坏性牙周病的主要风险因素,并深刻影响龈下微生物组,使其更具多样性、富含病原体而缺乏共生菌(Winkelhoff 等人,2001;Shchipkova 等人,2010;Bizzarro 等人,2013;Moon 等人,2015),即使在深度≤4毫米的浅牙周袋中也是如此(Haffajee 和 Socransky,2001)。吸烟者在牙周治疗后共生菌的重新定植受损,因为他们保持着促炎症的宿主表型(Joshi 等人,2014)。
甲硝唑干扰细菌的核酸合成。其抗菌谱覆盖厌氧菌,包括牙周病原体牙龈卟啉单胞菌、福赛坦纳菌和齿垢密螺旋体(Loesche 等人,1984)。阿莫西林抑制细菌细胞壁的合成,并与甲硝唑协同作用,减少与侵袭性牙周炎相关的伴放线聚集杆菌(van Winkelhoff 等人,1989)。目前,仅有少数研究探讨了在吸烟者牙周治疗中辅助使用阿莫西林和甲硝唑对微生物群落的影响(Matarazzo 等人,2008;Faveri 等人,2014)。然而,这些研究仅使用 DNA-DNA 棋盘法检测了预先定义且数量有限的细菌。所谓的"下一代测序"技术允许对微生物动态进行无假设且不受限制的观察,因为无需对细菌进行预选。迄今为止,尚无使用下一代测序技术研究吸烟者在接受含或不含抗生素的牙周治疗后微生物动态变化的研究。因此,本研究旨在利用 16S rDNA 扩增子下一代测序技术,比较机械性牙周治疗联合或不联合阿莫西林与甲硝唑辅助治疗,对吸烟并患有牙周炎的患者龈下微生物组的影响。
材料与方法
患者特征
本研究使用了来自 ABPARO 研究的样本,这是一项关于牙周治疗期间辅助使用抗生素效果的多中心、随机、双盲、平行组、安慰剂对照研究(ISRCTN:64254080,ClinicalTrials.gov NCT00707369)(Harks 等人,2015)。吸烟者定义为自我声明吸烟且使用吸烟分析仪(Bedfont-Smokerlyzer®,Bedfont,英国)检测一氧化碳(CO)浓度≥7 ppm 的人(Harks 等人,2015)。所有吸烟者接受相同的机械性牙周治疗。在 345 名符合方案的患者中,共有 71 名吸烟者。其中,9 名被诊断为重度(III 期),62 名为中度牙周炎(II 期)(Tonetti 等人,2018)。7 名重度患者和 27 名中度患者接受了辅助安慰剂治疗;2 名重度患者和 35 名中度患者接受了阿莫西林和甲硝唑辅助治疗(500 毫克/400 毫克,每日三次,持续 7 天)。由于重度吸烟患者的治疗组样本量小且不平衡,本研究仅纳入并报告了中度牙周炎吸烟患者的数据。为确保基线时具有最佳临床可比性的分组,将安慰剂组的 27 名中度牙周炎吸烟者与抗生素组 35 名中度患病吸烟者中的 27 名进行了匹配(Ho 等人,2011)。对这 54 名吸烟者在治疗前和治疗后 2 个月的时间点样本进行了分析。龈下样本采集自四个象限中各有的一颗患有牙周炎且探诊深度≥6毫米的牙齿,采集方法如前所述(Hagenfeld 等人,2018, 2019)。在每个位点,将一根无菌纸尖(ISO45,Roeko Dental,Langenau,德国)插入 10 秒,所有纸尖取出后收集于一个无菌收集管中。两次采样时间点的采样位点保持一致。本研究经明斯特大学医学伦理委员会批准(编号:2016-505-f-S)。
文库构建
使用 QiaAmp DNA-Mini 试剂盒(Qiagen,Hilden,德国)从吸烟者和非吸烟者的样本中分离并纯化细菌基因组DNA。分别通过琼脂糖凝胶电泳和 Nanodrop(Thermo Scientific,Silverside,美国)测量来检查DNA的完整性和纯度。使用 Qubit 2.0 荧光计配合 Qubit dsDNA BR 检测试剂盒(Thermo Fisher,Waltham,MA,美国)进行DNA定量。使用 KAPA HiFi Hot Start DNA Polymerase(Ready Mix,KAPA-Biosystems,Boston,MA,美国)进行两次PCR。在第一次PCR中,使用通用真细菌带尾标签双索引扩增引物(扩增子大小为 291 bp,对应 E. coli 位置 515 至 806)来扩增细菌 16S rRNA 基因的 V4 高变区(Klindworth 等人,2013)。引物序列如下:EMP515f: TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGGTGYCAGCMGCCGCGGTAA806_R: GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC其中 16S rDNA 特异性区域以粗体标出。在第二次PCR中,使用 Nextera XT Index Kit Version 2(Illumina,San Diego,CA,美国)连接样本特异性的"条形码"引物和接头序列,该试剂盒提供 96 种条形码组合。最多可将 96 个文库进行标准化并混合,用于 Illumina MiSeq 测序运行,使用 MiSeq Reagent Kit Version 2(Illumina)生成 250 个碱基对的双端读长。
五个阴性对照样本含有分子生物学级水,用于识别每个测序运行中潜在的外源性污染。此外,每个测序运行还加入了一个均衡的模拟样本,其中包含 23 个已知物种(对应 24 个 RSV),以进行运行间质量控制。模拟样本的组成见补充文件 1。模拟样本的 DNA 提取步骤如前所述进行,仅有一处修改:模拟物种的 DNA 分离使用 Qiagen Genomic Tips 20/G(Qiagen,Hilden,德国)进行。所有 23 个单物种样本均调整为每微升 10⁷ 个 16S rRNA 操纵子拷贝。随后,通过等量混合这些稀释液来创建均衡的模拟样本,其总拷贝数为每微升 10⁷ 个 16S rRNA 操纵子,即每个物种每微升含 4.3 × 10⁵ 个 16S rRNA 操纵子。
序列生成、引物和接头去除
使用 Illumina 的 MiSeq Control Software v.2.6.2.1 操作 MiSeq 仪器。Real-time Analysis Software v.1.18.54 进行图像分析、碱基识别以及为每个循环的每个碱基分配质量分数。MiSeq Reporter Software v.2.6.3 用于解复用、生成 FastQ 文件以及去除接头。使用 Cutadapt v.1.8.1(Martin,2011)去除测序引物,采用默认设置(即允许接头区域最大错误率为 10%)。这些经过修剪的读长已提交至 EMBL 欧洲生物信息研究所的欧洲核苷酸档案库(http://www.ebi.ac.uk/ena/),研究登录号为 PRJEB35812。
去噪与合并
使用 R v.3.6.1 和 RStudio v.1.1.463(R Development Core Team,2017)配合 DADA2 v.1.12.1(Callahan 等人,2016)进一步处理已去除接头和引物的 FastQ 文件。除非下文明确提及,否则均使用默认设置(最值得注意的是,在合并正向和反向读长时,允许重叠区域零错配)。首先,在 3' 端对正向和反向读长各修剪一个碱基,以去除测序过程中附带的、没有质量分数的末端碱基。其次,在 5' 端对读长进行修剪,以便在合并时两端未配对碱基最少(根据 E. coli 序列计算,正向读长修剪 22 个碱基,反向读长修剪 20 个碱基)。第三,允许每条读长最多出现两个预期错误,以过滤掉低质量读长。最后,在去噪过程中,将形成核糖体序列变异(RSV)分区所需的最小丰度设置为每个测序运行 10 条读长。
分类学标记与去污染
使用朴素贝叶斯分类器和 SILVA v.128 训练集(Pruesse 等人,2007)对 RSV 进行分类学标记。每个测序运行的结果都使用 phyloseq v.1.19.1(McMurdie 和 Holmes,2013)存储为一个单独文件,包含每个 RSV 的分类学标签和读长数。使用 decontam 包 v.1.4.0(Davis 等人,2018)对通过质量控制检查的每个测序运行进行独立筛查,以识别潜在的外源性污染物。采用结合"普遍性"和"频率"的方法,使用默认阈值 0.1 来识别污染物。随后,将所有单次运行的 phyloseq 文件合并,并与临床及人口统计学变量结合,生成最终的 phyloseq 文件。最后,对所有运行进行普遍性过滤,剔除仅在两个或更少样本中出现的 RSV,以去除假阳性结果。
测序运行与样本质量控制
在每个测序运行完成后,分两个连续步骤进行运行质量控制。首先,使用 MiSeq Reporter 软件检查运行的 Q30 分数。此处,该值必须高于制造商推荐的 75%(Illumina,2012)。其次,使用 DADA2 流程对模拟样本进行去噪处理,并用 phyloseq 进行检查。当一个运行中发现的 24 个 RSV 与模拟样本中 23 个物种的 16S RNA 参考基因完全匹配,且每个样本中未匹配到参考序列的读长数 ≤ 0.5% 时,即判定该运行质量合格。样本质量控制首先在每个运行完成后对所有样本进行,并在创建包含所有纳入运行的最终 phyloseq 文件后再次进行。若稀释曲线质量控制未显示饱和状态,或样本读长数 ≤ 5,000,则判定样本质量不合格。
统计分析
在群落水平分析中,使用 phyloseq 分析细菌多样性。对于 Alpha 多样性,确定了丰富度(即每个样本中随机二次抽样观察到的每个 RSV 的读长数)。此外,还提供了香农多样性指数。对于 Beta 多样性,创建了布雷-柯蒂斯距离矩阵,并计算了每位患者的平均相异性。分类变量的组间差异使用 Fisher 精确检验。所有其他连续变量的组间差异使用双侧 Mann-Whitney U 检验。对于两组内治疗前后连续变量的变化,则使用 Wilcoxon 符号秩检验。
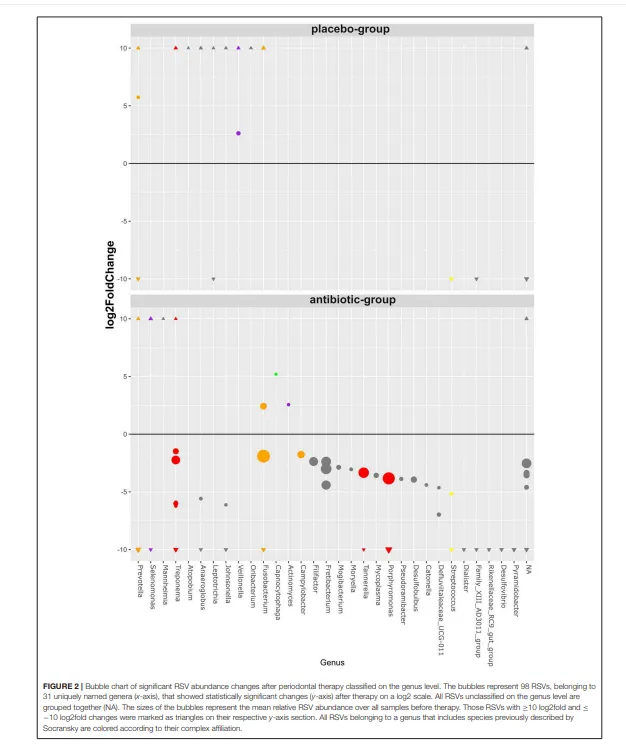
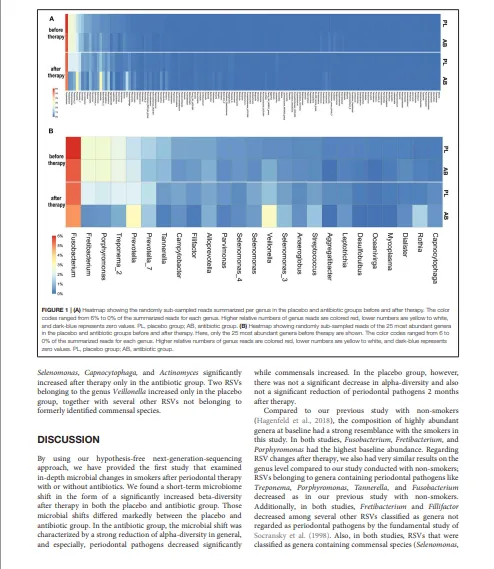
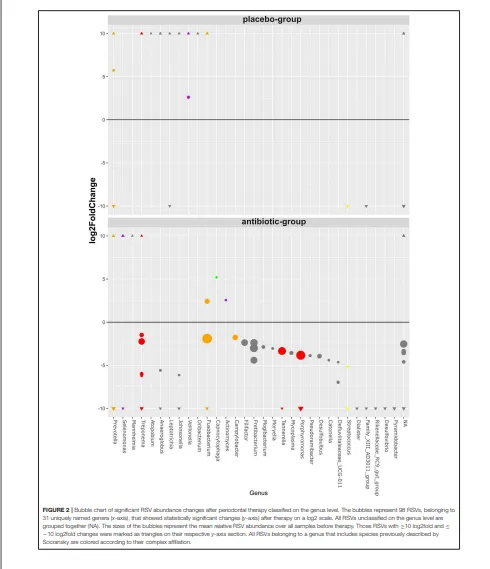
抗生素组和安慰剂组在治疗前后,每个菌属的随机二次抽样 RSV 平均读长数差异以热图形式展示。对于在菌属水平分类的单 RSV 差异丰度统计分析,使用 R 包 DESeq2 v.1.18.1(Love 等人,2014)。模型公式包含治疗组(AB,安慰剂)、时间点(前,后)以及两组间的交互项作为固定效应。将抗生素组和安慰剂组中发生显著变化的、按菌属分组的 RSV 在气泡图中展示。若一个菌属包含被 Socransky 小组(Socransky 等人,1998)认为与牙周疾病或健康相关的物种,则该菌属按先前描述的方法(Hagenfeld 等人,2018)归入相应的复合体。
结果
治疗前后的人口统计学、临床参数及测序输出预处理
治疗前,安慰剂组和抗生素组在所有检查的人口统计学和临床参数上均无统计学显著差异(表 1)。治疗后,两组的临床参数均显著改善(补充表 1,补充文件 2)。所有测序运行均通过了如前所述的运行质量控制(补充表 3)。总共发现 5 个 RSV 疑似外源性污染物并被移除。此外,有 645 个 RSV 在 ≤2 个样本中出现(普遍性低),同样被移除;同时移除的还有 2 个未被归类为细菌界的 RSV。所有样本在稀释曲线分析中均显示饱和,且每个样本读长数均超过 5,000(数据未显示)。
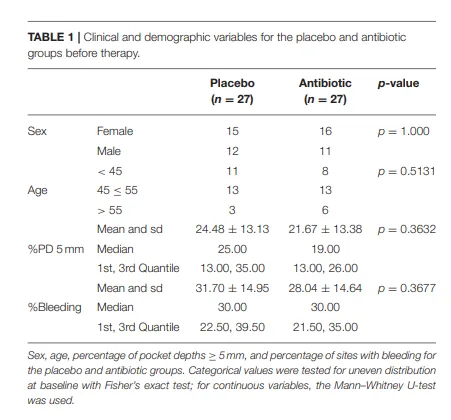
表1 | 治疗前安慰剂组与抗生素组的临床及人口统计学变量。
治疗前后的微生物多样性参数
治疗前,安慰剂组和抗生素组在所有测试的微生物多样性参数上均无统计学显著差异:平均丰富度(安慰剂组 161.81 ± 53.27 标准差 vs. 抗生素组 169.93 ± 57.32 标准差,p = 0.595)、平均多样性(安慰剂组 3.84 ± 0.39 vs. 抗生素组 3.86 ± 0.41,p = 0.797)和平均相异性(安慰剂组 3.84 ± 0.39 vs. 抗生素组 3.86 ± 0.41,p = 0.757)。治疗后,安慰剂组(p = 0.034)和抗生素组(p < 0.001)的相异性均显著增加。安慰剂组的丰富度(p = 0.768)和多样性(p = 0.090)未发生显著改变。然而,在抗生素组中,丰富度(p = 0.002)和多样性(p = 0.016)均显著下降。详细的多样性参数表可在补充表 2 中找到。
治疗前后的菌属水平丰度
我们共发现1060个RSV,可归属于143个不同的菌属(补充文件3)。治疗前,安慰剂组和抗生素组按菌属汇总的二次抽样读长相对数量高度相似(图1)。治疗前,抗生素组与安慰剂组之间,没有任何一个菌属的差异超过1.0%。治疗前差异最大的RSV属于梭杆菌属、韦荣球菌属和普雷沃菌属,其差异均低于0.5%。治疗前相对丰度最高的三个菌属是:梭杆菌属(安慰剂组:6.0% vs. 抗生素组:5.5%)、韦荣球菌属(安慰剂组:0.4% vs. 抗生素组:0.8%)和普雷沃菌属(安慰剂组:1.4% vs. 抗生素组:1.1%)。
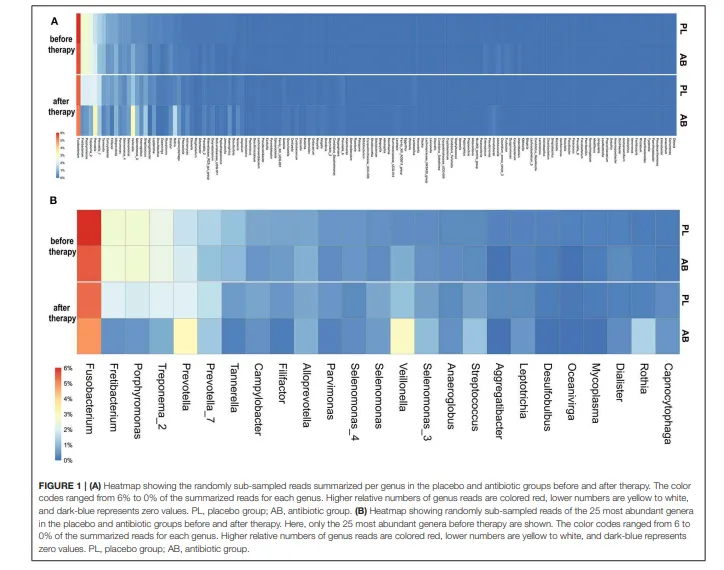
治疗后,安慰剂组与抗生素组的微生物组变得差异更大。治疗后差异最大的菌属是韦荣球菌属,其在两组中治疗后均有所增加,相对丰度在安慰剂组为1.1%,而在抗生素组为1.9%。纤毛菌属和卟啉单胞菌属在安慰剂组的减少幅度较小;它们的相对丰度在安慰剂组分别为2.0%和1.9%,而在抗生素组分别为0.4%和0.5%。在安慰剂组,没有任何一个菌属的相对丰度变化超过百分之一。在抗生素组,有六个菌属的变化超过百分之一。其中,三个菌属(纤毛菌属、卟啉单胞菌属和密螺旋体属)减少超过1.0%,三个菌属(韦荣球菌属、罗氏菌属和普雷沃菌属)增加超过1.0%。在安慰剂组,治疗后也主要是牙周病原菌减少而共生菌增加。然而,这些变化的幅度通常比抗生素组小约0.5–1.5%。
治疗后发生显著变化的、按菌属分组的RSV
治疗前,绝大多数(98.8%)RSV在抗生素组和安慰剂组之间的丰度没有统计学差异。治疗前发现的21个丰度有差异的RSV均属于低丰度RSV,其平均基线丰度低于每个样本的1%(补充文件4)。属于卟啉单胞菌属、坦纳菌属和密螺旋体属等包含牙周病原体菌属的RSV,其丰度仅在抗生素组显著下降,同时还有几个其他菌种也是如此(图2)。
讨论
通过采用我们这种无假设的高通量测序方法,我们首次深入研究了吸烟者在接受含或不含抗生素的牙周治疗后的微生物变化。我们发现,在安慰剂组和抗生素组中,治疗后均出现了短期微生物组转变,表现为β-多样性显著增加。然而,安慰剂组和抗生素组之间的微生物转变存在明显差异。在抗生素组,微生物转变的特征是α-多样性普遍大幅降低,特别是牙周病原菌显著减少而共生菌增加。然而在安慰剂组,α-多样性并未显著降低,治疗后两个月牙周病原菌也未显著减少。与我们之前针对非吸烟者的研究(Hagenfeld等人,2018)相比,基线时高丰度菌属的组成与本研究的吸烟者高度相似。两项研究中,基线丰度最高的菌属均为梭杆菌属、纤毛菌属和卟啉单胞菌属。关于治疗后的RSV变化,我们在菌属水平上的结果也与针对非吸烟者的研究非常相似:属于密螺旋体属、卟啉单胞菌属、坦纳菌属和梭杆菌属等包含牙周病原体的菌属的RSV减少了,这与我们之前对非吸烟者的研究结果一致。此外,两项研究中,纤毛菌属和丝状杆菌属都减少了,而这两个菌属以及其他几个被归类为Socransky等人(1998)基础研究中不视为牙周病原菌的属的RSV也减少了。同样,两项研究中,被归类为包含共生菌种(新月形单胞菌属、二氧化碳嗜纤维菌属和放线菌属)的菌属的RSV在抗生素组都有所增加。然而,在非吸烟者的安慰剂组中,我们没有发现相异性变化,而且在我们之前针对非吸烟者的研究中,安慰剂组只有三个RSV发生了显著变化。
先前一项使用DNA-DNA棋盘法的研究(Matarazzo等人,2008)与我们的研究具有很强的可比性,因为他们直接比较了两组接受不同牙周治疗的吸烟牙周炎患者:一组使用两种抗生素辅助治疗,另一组不使用抗生素。该研究中,接受阿莫西林和甲硝唑治疗的患者发生了与我们研究相似的微生物转变,同样仅在抗生素组中,卟啉单胞菌属、坦纳菌属和密螺旋体属等牙周病原菌属显著减少,并伴有放线菌属、新月形单胞菌属和二氧化碳嗜纤维菌属等共生菌种的增加。另一项使用DNA-DNA棋盘法的研究比较了阿莫西林和甲硝唑在吸烟者和非吸烟者中的使用效果。该研究中,接受辅助抗生素治疗的吸烟者同样出现了上述三个包含牙周病原菌的菌属的显著减少(Faveri等人,2014)。其他研究小组利用DNA-DNA棋盘法检查了吸烟者和非吸烟者接受不含抗生素的牙周治疗,发现与不吸烟者相比,吸烟者牙周病原菌的减少程度较低(Darby等人,2005;Feres等人,2015)。我们的研究结果也证实了这一点:安慰剂组中的吸烟者,其高丰度细菌的相对数量总体上略有减少,但治疗后没有牙周病原菌相关的RSV发生显著变化。
与这些先前基于假设的研究相比,在我们这项针对接受辅助抗生素治疗的吸烟患者的研究中,显著减少的菌属大部分(55%)不被视为牙周病原菌(Socransky等人,1998)。例如,未培养的纤毛菌属人类口腔分类单元360最近在日本人群中被发现是牙周炎的一种新型生物标志物(Khemwong等人,2019)。纤毛菌属的RSV在我们研究的德国吸烟人群中也高度富集,并在接受抗生素牙周治疗后显著减少。另一个例子是,被视为新出现的牙周病原体的福赛斯坦纳菌(Aruni等人,2014),以及从成人牙周炎患者牙周袋和感染根管中分离出的新菌属——穆氏菌属(Nakazawa等人,2000)。在我们的抗生素治疗研究中,这两个菌属都显著减少。正如最近一篇综述所指出的(Perez-Chaparro等人,2014),这些发现指出了这些以及其他先前未被识别的物种在牙周疾病治疗中的重要性。我们的研究也存在一些局限性。由于我们使用的250-bp读长限制,无法实现更低分类层级(如种水平)的更高分辨率分类。我们无法像宏基因组学方法那样评估微生物组的功能特性。此外,用于分析RSV水平变化的负二项回归模型无法考虑患者的随机效应。通过使用混合微生物样本,我们无法评估位点特异性差异。我们仅纳入了中度牙周炎患者。要评估治疗后变化的稳定性和重新定植的模式,还需要更长时间的随访。
结论
通过对吸烟的牙周炎患者进行无假设的16S rDNA扩增子高通量测序研究,我们发现,属于包含牙周病原菌的菌属的RSV仅在辅助使用阿莫西林和甲硝唑时才减少。此外,我们扩展了在吸烟者中,牙周治疗后短期内发生变化的细菌谱,范围超出了已被深入研究的牙周病原菌和共生菌。





声明:本文翻译自国外病例展示,仅供口腔专业人士进行技术交流,仅代表医生个人观点,不构成任何医疗建议,如有翻译错误之处敬请指正。
END

微信公众号:chi-xue-tang
扫码关注 获得更多精彩
本公司对外合作联系:18361831107
投稿邮箱:chixuetang@163.com