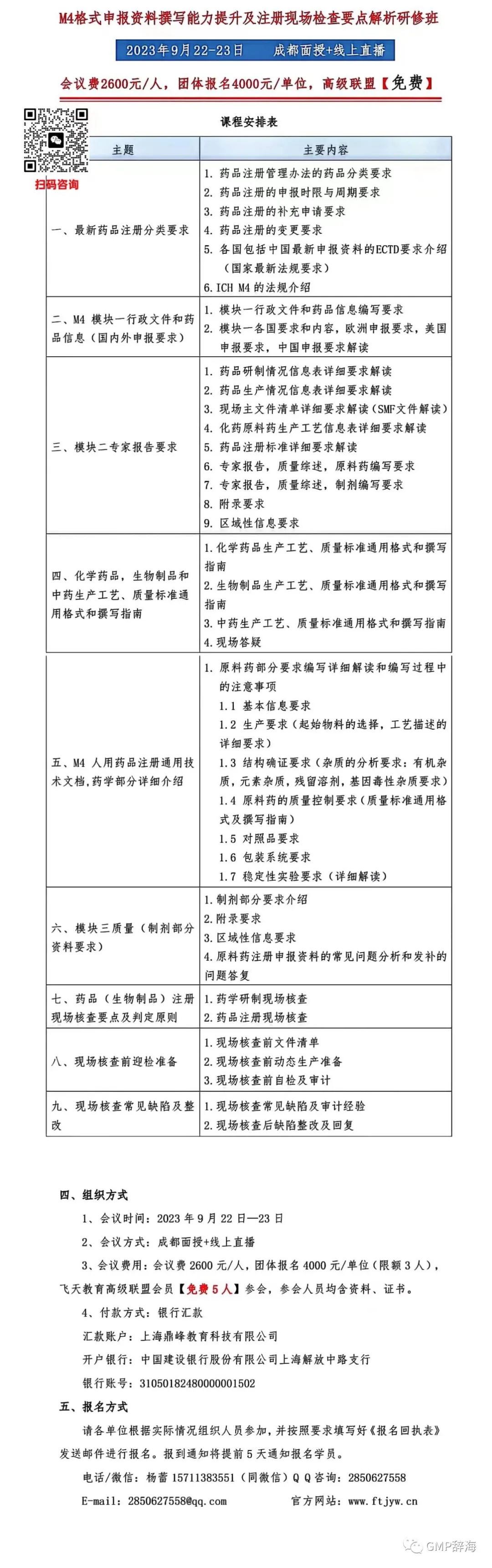
我们首先来看第一封警告信中关于清洁验证的内容。
During their inspection, FDA investigators found "spalling" of the inner surface and incrustations on equipment components. Furthermore, residues were also found on an inner surface of one piece of equipment.
The manufacturer stated that the residues remaining were non-significant and non-reactive. Furthermore, the manufacturer wanted to implement preventive measures regarding the update of a work instruction and regarding the handling of the "spalled" material. Additionally, a verification checkpoint is to be established.
FDA's responses in the Warning Letter to the inspected company's replies that this is not sufficient for the FDA! The FDA demands an evaluation of the potential risk of "spalling" for all APIs that are manufactured there.
A summary, retrospective review of cleaning effectiveness with respect to cross-contamination risks. The identities of residues, equipment other than those previously considered that may also have been inadequately cleaned, and a consideration of whether cross-contaminated products may have been released are to be included. The consideration should include any inadequate cleaning procedures and practices and any equipment item in which more than one product is manufactured.
Based on this retrospective review, a CAPA plan shall then be developed regarding the cleaning programme. This should identify corrective actions to the cleaning process and activities and provide timelines to implementation. Furthermore, the FDA would like to see a summary of weaknesses in the life cycle management of cleaning. Improvements to the cleaning programme, including improvements to cleaning efficiency and improved continued verification of cleaning execution of all products and equipment, should be described. The cleaning validation programme should be improved to consider worst case scenarios, regarding:
毒性较高的药品。
2. Medicinal products with higher API contents.
原料药含量较高的药品。
在清洁试剂中溶解度低的药品。
具有难以清洁特性的药品。
用棉签擦拭采样的最难清洁的位置。
清洗前的静置时间。
当新设备或新产品引入时,对变更管理系统的调整。
更新SOP的概述,以确保产品、工艺和设备有足够的清洁过程的确认和验证程序。
对每台设备的清洁操作和相关的清洁验证策略进行全面审查,以确定是否存在类似的缺陷。
以下是第二封警告信中关于清洁验证的内容。
As in the November 2022 inspection, the issue of cross-contamination was addressed again, this time also mentioning the relevant passage from the Code of Federal Regulations (CFR), 21 CFR 211.67 (a). Residues were once more found on interior surfaces, but this time turned out to contain APIs. The company admitted that some areas had not been cleaned or inspected for contaminants since installation 14 years ago. A placebo batch produced during the inspection also showed contamination with API.
In its response to the actions listed above, the FDA first and foremost criticized the company's conclusions about the placebo batches with no detectable contamination. The reason: cross-contamination is not "equally distributed." And citing non-contaminated placebo batches as justification for appropriate cleaning of visibly contaminated equipment is unscientific. In addition, recovery rates after performing the placebo batches mostly occurred on surfaces not in contact with the product.
Besides to the placebo batches, of course, the FDA then criticized the fact that recovery rates were taken at locations that did not necessarily contain the highest levels of contamination and the sampling method itself. The same more extensive measures were called for as were named in the Warning Letter following the 2022 inspection at the other site.
Conclusion: "Simple responses" to inspection deficiencies to the FDA are often inadequate. FDA typically wants to see that the deficiencies found are evaluated retrospectively and described proactively as to what corrective actions will prevent the deficiencies in the future (CAPA). The FDA urges a scientific approach to cleaning validation here. Their Guide to Inspection Validation of Cleaning explicitly mentions "swab" and "rinse" as sampling techniques, not placebo batches.
结论:对FDA检查缺陷的“简单回应”往往是不够的。FDA通常希望看到对发现的缺陷进行回顾性评估,并主动描述哪些纠正措施将防止未来的缺陷。FDA敦促采用科学的方法进行清洁验证。他们的《清洁检验验证指南》明确提到“拭子”和“漂洗”作为取样技术,而不是通过安慰剂批次来证明清洁的有效性。
免责声明:本公众号所载文章版权归原作者所有。转载文章旨在知识分享,如涉及作品内容、版权和其它问题,请联系我们删除!文章内容为作者个人观点,并不代表本公众号赞同或支持其观点。本公众号拥有对此声明的最终解释权。
请转发相关同事参考
欢迎转发关注公众号:GMP辞海每天都有干货内容分享!