


密度泛函理论(Density Functional Theory, DFT)作为现代凝聚态物理、量子化学及计算材料科学的基石,为人类在原子与电子尺度上理解物质结构与物理化学性质提供了极为关键的理论框架。自其建立以来,DFT通过将高维且计算呈指数级复杂的全电子波函数多体薛定谔方程,严格映射为仅依赖于三维空间电子密度的单体Kohn-Sham方程,极大地降低了电子结构计算的自由度与计算开销。本报告基于理论物理与计算化学的客观数据与历史文献,详尽剖析密度泛函理论的历史奠基、主流算法生态体系的演变、现存理论的本征物理误差,以及当前在无轨道形式、深度学习神经网络泛函与量子计算混合架构等前沿领域的范式重构。
一、 密度泛函理论的物理溯源与数学架构确立
1.1 从多体波函数到电子密度模型的初期探索
量子力学建立初期,1926年薛定谔方程的提出为微观体系的描述提供了严格的数学基础。然而,正如保罗·狄拉克所指出的,基于严格量子力学基本定律推导宏观化学行为的数学方程过于复杂,在处理实际多电子体系时面临“指数墙”效应,即求解包含 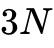 个空间变量的多体波函数在计算上是不可行的。在此背景下,1927年建立的Thomas-Fermi (TF) 模型成为人类首次尝试将多体系统简化为电子密度函数的开创性工作。TF模型以及随后引入交换作用的Thomas-Fermi-Dirac (TFD) 模型假设,多电子系统的动能和交换能可以通过均匀电子气模型的局部能量密度进行近似计算。尽管该模型在理论上具有启发性,但由于其彻底忽略了电子壳层结构的量子效应且无法正确描述化学键的形成,在分子系统和精确化学应用中被证明是失效的。与此同时,基于斯莱特行列式(Slater determinant)的Hartree-Fock (HF) 理论虽然通过非局域交换算符满足了泡利不相容原理,但彻底忽略了电子间的动态相关效应(Dynamic correlation),同样无法达到化学精度。
个空间变量的多体波函数在计算上是不可行的。在此背景下,1927年建立的Thomas-Fermi (TF) 模型成为人类首次尝试将多体系统简化为电子密度函数的开创性工作。TF模型以及随后引入交换作用的Thomas-Fermi-Dirac (TFD) 模型假设,多电子系统的动能和交换能可以通过均匀电子气模型的局部能量密度进行近似计算。尽管该模型在理论上具有启发性,但由于其彻底忽略了电子壳层结构的量子效应且无法正确描述化学键的形成,在分子系统和精确化学应用中被证明是失效的。与此同时,基于斯莱特行列式(Slater determinant)的Hartree-Fock (HF) 理论虽然通过非局域交换算符满足了泡利不相容原理,但彻底忽略了电子间的动态相关效应(Dynamic correlation),同样无法达到化学精度。
1.2 现代DFT的理论奠基:Hohenberg-Kohn定理与Kohn-Sham方程
现代密度泛函理论的确立归功于20世纪60年代中期的两项底层数学证明与物理重构。1964年,Pierre Hohenberg与Walter Kohn发表了奠基性的HK定理,在数学上严格证明了对于处于非简并基态的多电子体系,其所受的外部势场(进而决定系统的完整哈密顿量及所有基态性质,包括总能量)唯一地由其基态电子密度 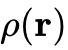 决定。HK定理将系统的复杂性从
决定。HK定理将系统的复杂性从 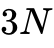 维的波函数降维至三维空间的密度分布,确立了电子密度的核心物理地位。
维的波函数降维至三维空间的密度分布,确立了电子密度的核心物理地位。
然而,HK定理仅证明了密度泛函的存在性,并未给出具体的动能泛函形式。传统的OF-DFT由于无法精确表达动能泛函而长期受限。为了克服这一障碍,1965年,Kohn与Sham提出将真实的相互作用多体系统映射为一个密度完全相同的、无相互作用的虚拟参考系统(即Kohn-Sham体系)。这一重构使得系统的绝大部分动能可以通过引入一组单电子辅助轨道(Kohn-Sham轨道)进行极其精确的解析计算。真实系统动能与无相互作用系统动能之间的微小差值,连同非经典电子间排斥作用(交换与相关效应),被统一归集于一个未知的交换相关泛函(Exchange-Correlation Functional, 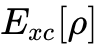 )中。Kohn-Sham方程的建立标志着DFT从纯粹的数学存在性定理转化为具有极强工程与科研实用价值的自洽场计算框架。
)中。Kohn-Sham方程的建立标志着DFT从纯粹的数学存在性定理转化为具有极强工程与科研实用价值的自洽场计算框架。
1.3 交换相关泛函的演进与“雅各布天梯”
由于确切的 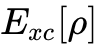 数学形式至今仍是未知数,过去几十年DFT的发展史实质上是物理学家与化学家对这一泛函不断逼近与近似改进的历史。Tulane大学对引文数据的分析显示,DFT是1980至2010年三十年间物理学中最活跃的领域,顶级物理学家如Perdew和Becke的单篇引用量高达数万次,彰显了该理论的深远影响。理论物理学家John Perdew于2001年提出了著名的“雅各布天梯”(Jacob's Ladder)分类体系,根据泛函中引入的微观物理信息维度,将现有的近似泛函划分为五个递进的阶梯:
数学形式至今仍是未知数,过去几十年DFT的发展史实质上是物理学家与化学家对这一泛函不断逼近与近似改进的历史。Tulane大学对引文数据的分析显示,DFT是1980至2010年三十年间物理学中最活跃的领域,顶级物理学家如Perdew和Becke的单篇引用量高达数万次,彰显了该理论的深远影响。理论物理学家John Perdew于2001年提出了著名的“雅各布天梯”(Jacob's Ladder)分类体系,根据泛函中引入的微观物理信息维度,将现有的近似泛函划分为五个递进的阶梯:
局域密度近似(LDA):天梯的第一层,其交换相关能量仅依赖于该空间点的局域电子密度
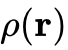 。LDA从均匀电子气模型演化而来,在描述简单的碱金属等离域固态系统时表现尚可,但由于存在严重的自相互作用误差且无法反映密度的空间不均匀性,对分子解离能等化学性质的预测完全失效。
。LDA从均匀电子气模型演化而来,在描述简单的碱金属等离域固态系统时表现尚可,但由于存在严重的自相互作用误差且无法反映密度的空间不均匀性,对分子解离能等化学性质的预测完全失效。广义梯度近似(GGA):天梯的第二层,不仅包含电子密度本身,还引入了密度的梯度
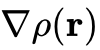 ,从而能够部分捕捉电子密度的非均匀特征(代表性泛函包括PBE、PW91)。GGA在20世纪80年代促使DFT真正进入化学研究领域,大幅提高了分子几何构型和内聚能的预测精度。
,从而能够部分捕捉电子密度的非均匀特征(代表性泛函包括PBE、PW91)。GGA在20世纪80年代促使DFT真正进入化学研究领域,大幅提高了分子几何构型和内聚能的预测精度。元广义梯度近似(meta-GGA):天梯的第三层,进一步引入了动能密度
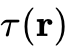 或电子密度的拉普拉斯算符
或电子密度的拉普拉斯算符 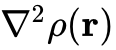 (代表性泛函如TPSS、SCAN)。该层级泛函在处理固体表面性质和部分弱相互作用时展现出比常规GGA更高的精度与数值稳定性。
(代表性泛函如TPSS、SCAN)。该层级泛函在处理固体表面性质和部分弱相互作用时展现出比常规GGA更高的精度与数值稳定性。杂化泛函(Hybrid Functionals):天梯的第四层,将一定比例的Hartree-Fock精确交换能与GGA或meta-GGA交换相关能进行线性混合(代表性泛函包括B3LYP、PBE0、HSE06)。精确交换项的引入在很大程度上抵消了半局域泛函固有的自相互作用误差,显著提升了半导体带隙、化学反应势垒和各类热化学性质的绝对准确度。
完全非局域与多体方法:天梯的最高层,引入了未占位轨道信息或依赖于轨道系统的非局域响应(如无规相近似RPA、双杂化泛函)。这些方法理论精度极高,但也伴随着极大的计算开销,通常被局限在小分子基准测试之中。
长期以来,这套自下而上的发展体系面临着一个无法逃避的物理权衡(Trade-off):泛函在天梯上的每一次攀升虽然提高了预测精度,但其渐近计算成本也随之呈指数或高阶多项式级别增加,阻碍了精确电子结构方法在大规模凝聚态系统中的应用。
二、 主流软件架构、硬件加速与计算标度的演变
随着计算材料科学的爆发式增长,底层波函数展开基组的选择决定了各类DFT软件框架的计算效率、资源消耗及适用场景,并逐步分化出适应不同硬件环境的代码生态。
2.1 主流DFT代码与基组依赖特性
当前的计算生态中,由于分子体系和周期性晶体体系边界条件的不同,软件工具呈现出高度特化的发展趋势。平面波、高斯函数以及局域轨道基组的选择直接影响了求解哈密顿矩阵和泊松方程的数值策略。
软件名称 | 主要适用目标体系 | 核心底层算法与基组特征分析 | 硬件与生态地位 |
VASP | 周期性固体 / 表面物理 | 采用平面波基组与PAW(投影缀加波)赝势,精确处理核心电子。 | 固态材料计算的工业级标准,高度优化。 |
Quantum ESPRESSO | 周期性固体 / 纳米材料 | 平面波基组结合超软赝势;在线性响应理论和声子计算方面功能全面。 | 免费开源;已完成向百亿亿次(Exascale)GPU硬件的高效移植。 |
Gaussian | 离散分子体系 / 化学反应 | 基于原子中心的高斯型轨道基组,对处理非周期性边界条件极其高效。 | 传统化学工业标准计算工具;提供全面的GUI与构象搜索支持。 |
ORCA | 分子体系 / 过渡金属配合物 | 局域化基组与分辨率恒等(RI)近似技术结合,显著加速库仑和交换积分。 | 免费提供学术使用;在双杂化泛函、旋轨耦合及高阶多参考计算中速度优势显著。 |
异构计算与GPU加速的深度基准测试
现代超级计算机正向GPU主导的异构架构演进。历史上的GPU版DFT算法在处理高角动量基函数时常面临严重效率瓶颈,使得它们在需要大基组的高精度能量计算中作用受限。然而,最新一代基于GPU的量子化学框架(如 GPU4PySCF)展现了革命性的标度行为。基准测试表明,当针对直链烷烃类系统采用含极化和弥散函数的基组(如 def2-TZVP 或 def2-QZVP,其中碳原子包含 f 或 g 角动量函数)计算 r2SCAN 能量时,GPU算法突破了传统CPU瓶颈。测试呈现出两种显著的缩放机制:针对小体系的“过剩显存”区间与针对大体系的“批处理优化”区间。例如,在应用具有更大显存配置的硬件(如从A100-40GB向A100-80GB迁移)时,对于未能饱和显存的较小分子,性能提升微弱;但对于大规模复杂系统,扩大显存结合批处理调度能引致阶跃性的算力跃升,从而为大规模材料筛选奠定了硬件基础。
2.2 线性标度(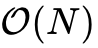 )方法的物理原理与软件实现
)方法的物理原理与软件实现
传统Kohn-Sham DFT的计算成本随原子总数 的增长呈 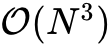 指数式膨胀,这一标度瓶颈主要源自求解本征值问题时全局哈密顿量矩阵的直接对角化过程以及施加分子轨道正交性约束所需的操作。因此,常规计算通常被严格限制在数百至数千原子的范围。为了突破这一极限,理论界基于Kohn在1996年提出的“电子物质的近视原理”(Nearsightedness of electronic matter),发展了大量线性标度(
指数式膨胀,这一标度瓶颈主要源自求解本征值问题时全局哈密顿量矩阵的直接对角化过程以及施加分子轨道正交性约束所需的操作。因此,常规计算通常被严格限制在数百至数千原子的范围。为了突破这一极限,理论界基于Kohn在1996年提出的“电子物质的近视原理”(Nearsightedness of electronic matter),发展了大量线性标度(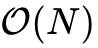 )方法。该原理指出,宏观体系中空间两点间的一阶密度矩阵随着空间距离的增加呈现出指数级的衰减规律。
)方法。该原理指出,宏观体系中空间两点间的一阶密度矩阵随着空间距离的增加呈现出指数级的衰减规律。
基于上述原理的线性标度方法直接避免了求解全局扩展波函数,转而采用局域化函数表示单粒子密度矩阵,从而将计算复杂度降维至 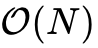 。
。
ONETEP程序:采用了一组严格局域化的非正交广义Wannier函数(NGWFs)作为基函数,并通过一种被称为 psinc(周期性sinc函数)的基础网格实现表达。ONETEP的算法优势在于其既具备了等价于平面波基组的高精度特性,又通过分布式的密度矩阵优化避免了昂贵的库仑求和,使得计算规模与内存消耗随原子数量呈线性增长。测试表明,其在包含数万原子的非均匀系统中表现出极佳的MPI并行效率。
CONQUEST程序:这一大型代码被设计用于百亿亿次级别的超算平台,能够同时执行精确对角化和线性标度算法。其底层采用了两种局域基组:伪原子轨道(PAOs)与系统收敛的B样条函数。在采用多位点支持函数(MSSF)的情况下,能够以极高的精度模拟含上万个原子的复杂体系。在大规模线性标度基准测试中,CONQUEST已被证明能够处理超过一百万个原子,并且在超级计算机(如Fugaku和K computer)上能够完美展现弱缩放特性(固定每核心原子数下维持恒定计算时间),调度多达20万个处理器核心协同工作。
BigDFT与金属体系的突破:需要指出的是,传统的依赖近视原理的
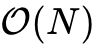 算法(包括直接变分或净化方法)通常假设体系存在有限的电子带隙。对于密度矩阵局域化急剧弱化或失效的金属及强离域系统,这一前提往往不成立。近年来,基于光谱正交化(SQ)以及无需依赖近视原理约束的无正则分解(iPEXSI)算法逐步融入现有的低标度软件生态中,利用矩阵的三角分解局域化性质,在非零电子温度条件甚至零温下,成功实现了对铝等典型金属原子的亚线性至线性计算扩展。
算法(包括直接变分或净化方法)通常假设体系存在有限的电子带隙。对于密度矩阵局域化急剧弱化或失效的金属及强离域系统,这一前提往往不成立。近年来,基于光谱正交化(SQ)以及无需依赖近视原理约束的无正则分解(iPEXSI)算法逐步融入现有的低标度软件生态中,利用矩阵的三角分解局域化性质,在非零电子温度条件甚至零温下,成功实现了对铝等典型金属原子的亚线性至线性计算扩展。
三、 高通量计算平台、材料数据库与可重复性挑战
随着DFT底层计算算法的成熟,由超级计算机集群驱动的高通量计算(HT-DFT)彻底改变了材料发现的范式。通过系统性地扫描海量的成分与结构组合,计算材料学界已建立起一系列庞大的数据基础设施,用于加速新能源电池、催化剂以及半导体材料的发现进程。
数据库生态体系 | 核心数据规模与覆盖特征 | 应用与接口特征 |
Materials Project (MP) | 依托NERSC超算,涵盖超过15万种无机化合物。 | 提供包括能带结构、弹性模量、压电张量及点缺陷性质的深度数据,具备极强的API生态。 |
OQMD (开放量子材料数据库) | 包含约30万个DFT总能量基态和低能虚构装饰结构的记录。 | 聚焦形成能与热力学稳定性分析,预测了超3200种未经实验表征的新型化合物。 |
AFLOW | 高通量合金筛选平台,收录数百万材料物性。 | 高度自动化,具有极强的对称性分析、多元相图生成与热力学稳定性自动化筛选功能。 |
NOMAD | 全球最大的独立且互联第一性原理数据平台。 | 收录超过1200万个晶体与分子数据,严格遵守FAIR原则,可跨不同DFT软件(如VASP,FHI-aims)检索计算源数据。 |
数据异质性与跨库可重复性危机
尽管高通量数据库极大促进了机器学习势函数(MLFFs)和性质预测AI(如Matbench平台)的繁荣,但HT-DFT研究中存在一个不可忽视的隐患:不同平台间基态性质的计算再现性极具挑战。一项汇编了AFLOW、Materials Project以及OQMD中大量同构晶体数据记录的研究揭示了严重的底层参数发散效应。通过对比数据库中起始晶体结构完全一致的条目发现:系统形成能和晶胞体积表现出相对良好的一致性,形成能的平均绝对相对差异(MRAD)为 6%(最大偏差达 0.105 eV/atom),体积 MRAD 仅为 4%。
然而,针对更高阶的电子特征属性,分歧被急剧放大。不同数据库计算出的带隙绝对差异达到 0.21 eV(MRAD 9%),而在总体磁化强度预测方面,各库计算的磁矩相差达到 0.15 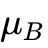 /单元(MRAD 8%)。更引人深思的是,由于高通量工作流对输入参数缺乏强制统一标准,高达7%的计算记录对于某一具体材料究竟呈现金属性还是半导体性存在定性分歧;在磁学判定上,15%的记录无法就某一结构是否具有自发磁化达成共识。这种差异的物理根源不仅在于波函数基底不同,更在于各数据库对核心区域所使用赝势的柔度设定不同,以及对于处理过渡金属局域
/单元(MRAD 8%)。更引人深思的是,由于高通量工作流对输入参数缺乏强制统一标准,高达7%的计算记录对于某一具体材料究竟呈现金属性还是半导体性存在定性分歧;在磁学判定上,15%的记录无法就某一结构是否具有自发磁化达成共识。这种差异的物理根源不仅在于波函数基底不同,更在于各数据库对核心区域所使用赝势的柔度设定不同,以及对于处理过渡金属局域 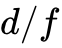 电子时采用的GGA+ 框架内有效哈伯德参数(值)设定的系统性偏差。这提示未来的大规模AI模型构建必须考虑到这些内置的方法学噪音。
电子时采用的GGA+ 框架内有效哈伯德参数(值)设定的系统性偏差。这提示未来的大规模AI模型构建必须考虑到这些内置的方法学噪音。
四、 DFT的深层物理局限:自相互作用与电子相关灾难
如果我们仅仅聚焦于计算规模的暴力扩展和数据库的积累,将无法掩盖DFT由于其基础近似所带来的内源性物理偏差。这些偏差深刻限制了DFT在强相关材料和精确光物理领域的应用。理解这些系统性失败,是推动泛函底层理论演化的必要前提。
4.1 自相互作用误差(SIE)与密度离域灾难
自相互作用误差(Self-Interaction Error, SIE)是当前半局域泛函面临的最普遍、最具破坏性的缺陷。在精确的多体量子力学理论中,单电子系统(如孤立氢原子)的经典库仑排斥能(Hartree能)应当与系统的纯交换能完美抵消,因为单一电子不可能与自身的电荷云发生静电排斥。但在目前基于LDA或GGA平均场框架的密度泛函近似中,非局域交换泛函的近似不足以完全抵消Hartree项中产生的这种虚假排斥。
为了降低这种无物理意义的自我排斥能,半局域泛函往往会错误地将电子密度向整个空间过度扩展,导致所谓的人为“离域误差”(Delocalization error)。这种物理特性的歪曲在面临拉伸的奇电子体系时表现得最为致命。例如,在计算最基本的 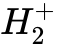 分子离子解离势能面时,随着两个质子距离的拉大,真实的单电子应该以相等的概率定域在某一侧质子上。但所有传统的GGA类泛函都会强制单电子在整个空间无限期地均匀分布,最终导致解离极限的预测能量偏离实际值高达 50-60 kcal/mol。自相互作用误差同样是导致化学反应势垒高度被系统性低估(人为稳定过渡态的过度离域电荷)的核心诱因。
分子离子解离势能面时,随着两个质子距离的拉大,真实的单电子应该以相等的概率定域在某一侧质子上。但所有传统的GGA类泛函都会强制单电子在整个空间无限期地均匀分布,最终导致解离极限的预测能量偏离实际值高达 50-60 kcal/mol。自相互作用误差同样是导致化学反应势垒高度被系统性低估(人为稳定过渡态的过度离域电荷)的核心诱因。
4.2 导数不连续性与固体带隙的系统性低估
凝聚态物理学界长期使用DFT进行能带结构计算,但遇到一个广为人知的严峻挑战:标准半局域泛函会极其严重地低估半导体及绝缘体的基本带隙,误差幅度通常高达30%至50%。具体案例表明,对于工业界极为关键的材料,如Si、GaAs、ZnO和GaN,采用PBE或LDA得到的带隙无法直接用于光电性质分析。
这种带隙低估并非由于Kohn-Sham单电子能级不具备物理意义,而是由精确交换相关能量泛函对电子占位数量的非解析行为所决定的。严格的理论推导指出,真实物理系统的基态能量 相对于系统的电子总数 必须是一条折线段。当系统电子数刚好经过一个整数临界点时,能量对于电子数的导数(本质上是化学势)会发生一次突然的非连续跳跃,这被称为导数不连续性(Derivative Discontinuity)。而当前广泛采用的LDA/GGA能量泛函在分数占位区是平滑可导的凸函数曲线,完全丢失了这种跳跃行为,最终造成带隙计算值缩水。
为了弥补这一基础缺陷,理论学家采取了不同层级的妥协手段:
杂化泛函(如 HSE06):通过在短程相互作用区间混入一定比例的Hartree-Fock精确交换作用,杂化泛函实质上人为引入了部分导数不连续特性。这种方法在抵消价带顶层自相互作用误差的同时,通过抬升导带底的能量,使得预测的带隙大幅逼近实验值(对于氮化物材料,HSE06的平均绝对百分比误差可降至11.1%)。
准粒子多体微扰理论(
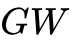 势):利用格林函数与屏蔽库仑势来严格描述激发态的电子增添/移除能。尽管
势):利用格林函数与屏蔽库仑势来严格描述激发态的电子增添/移除能。尽管 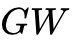 计算能够给出目前最高精度的半导体能带预测,但其极度高昂的计算成本使其在含有表面缺陷或大晶胞的超复杂体系中难以立足。
计算能够给出目前最高精度的半导体能带预测,但其极度高昂的计算成本使其在含有表面缺陷或大晶胞的超复杂体系中难以立足。
4.3 强相关系统的静态相关误差与DFT的边界
强相关材料(如含有部分填充 或 电子壳层的过渡金属氧化物体系、镧系及锕系元素)长期被认为是传统DFT失效的重灾区。在强相关物理图景下,电子间的动态库仑排斥占据主导地位,导致系统的真实基态波函数呈现高度纠缠的多组态性质,单一行列式的Kohn-Sham辅助系统假设彻底崩溃。
此类体系的计算误差被归类为静态相关误差(Static correlation error)。一个典型的失败测试模型是无限拉伸的无自旋中性 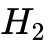 分子:在解离极限下,由于泛函无法准确重现局部自旋对称性的破缺与分数自旋态的稳定性能量,所有的现有半局域泛函都无法给出正确的解离行为。在工业研究中,常通过引入经验性的Hubbard 参数(DFT+ 或 SCAN+ 方法)增加对局域轨道的排斥惩罚以强制推开电子,但这破坏了第一性原理的客观普适性,也暴露出DFT在处理高阶多体作用时的固有脆弱性。
分子:在解离极限下,由于泛函无法准确重现局部自旋对称性的破缺与分数自旋态的稳定性能量,所有的现有半局域泛函都无法给出正确的解离行为。在工业研究中,常通过引入经验性的Hubbard 参数(DFT+ 或 SCAN+ 方法)增加对局域轨道的排斥惩罚以强制推开电子,但这破坏了第一性原理的客观普适性,也暴露出DFT在处理高阶多体作用时的固有脆弱性。
五、 激发态建模与光化学反应过程的困境与突围
除了基态属性,光激发与电子弛豫过程构成了太阳能捕获、光催化以及半导体发光机制的底层逻辑。含时密度泛函理论(TDDFT)利用Runge-Gross定理拓展了原有架构,成为目前最为广泛的激发态性质计算平台。然而,当面临更为复杂的瞬态光化学反应时,基于线性响应框架(LR-TDDFT)的常规算法存在诸多致命盲区。
5.1 传统线性响应TDDFT的瓶颈
传统的LR-TDDFT其响应空间本质上被限制在单电子激发构型的组合内。这一数学框架的天然缺陷导致其在以下物理场景中经常输出定性的错误结论:
电荷转移态(Charge-transfer states)低估:当电子供体与受体在空间上相距较远时,激发电子与空穴间的库仑束缚急剧减弱,而常用泛函无法捕捉长程渐近电位的正确衰减形式(即无法维持 行为),导致预测的电荷转移能态偏低。
核心能级激发(Core-level excitations)的能量偏移:在模拟X射线吸收光谱时,高能射线导致的内核电子跃迁伴随着剧烈的轨道弛豫效应,单次响应计算无法表征深层势垒的变动。
彻底丢失双激发态(Double excitations):这限制了模型在处理多烯烃视觉蛋白发色团或复杂过渡金属发光配合物时的适用性。
5.2 状态特异性演进与多参考自旋翻转TDDFT的崛起
为了绕开这些限制,计算化学界在近几年提出了多条具有突破性的技术路径以弥补TDDFT的短板:
态相关轨道优化密度泛函(State-Specific Orbital-Optimized DFT, OO-DFT):与依赖响应理论推演的方法不同,OO-DFT直接绕过基态参考框架,利用变分原理针对特定的高能激发态构型单独重新优化全套Kohn-Sham轨道。这种非正交性处理机制允许高激发系统(如核心电子耗尽态)拥有充分的密度弛豫自由度,在解决X光光谱学及高能物理问题时极具实用价值。
多参考自旋翻转TDDFT(MRSF-TDDFT):这是在2024-2025年取得重大影响的另一项新理论。该方法建立在一个创新的双重参考态基准之上,能够将电子激发与高阶组态相互作用在一个对称一致的基础上合并处理。更关键的是,MRSF-TDDFT自动引入了双激发组态进入其响应计算子空间,使其不仅能够在处理复杂的开壳层单线态体系(如双自由基化学,Diradicals)时给出精确判断,还能保证分子处于光反应中最极端的断键区域——即势能面圆锥交叉点(Conical Intersections)——依然保持完全正确的拓扑结构描述,计算出准确的非绝热耦合矢量,并取得了媲美计算量极大耦合簇理论的绝热单-三重态间距预测精度。
六、 物理法则与纯数据的博弈:机器学习泛函(ML-DFT)时代的降临
近几十年来,理论化学家始终在通过引入更加复杂的密度物理变量或精确交换算符来手工雕琢新的交换相关泛函结构。然而,受限于复杂的微积分约束与渐进标度,这种基于解析推导的方法被认为已接近天花板。自2021年起,机器学习(尤其是深度神经网络)的介入从根本上震撼了物理学界坚守的发展范式,试图通过直接映射超大规模波函数数据,实现精度与算力的解耦。需要明确区分的是,此类研究针对的是第一性原理底层的机器学习交换相关泛函(ML Exchange-Correlation Functionals),而非简单的基于拟合经典势能面的机器学习力场(MLFFs)。
6.1 DeepMind DM21 的突破及其数值振荡隐患
2021年,Google DeepMind提出了一种引起广泛关注的神经网络泛函——DM21模型。有别于传统的能量密度拟合,DM21团队巧妙地将含有分数电荷与分数自旋的病态物理约束(用于抑制前文所述的离域误差和静态相关系统崩溃)显式地注入神经网络的训练集中。DM21在解释诸如二聚体解离势能面和电荷强离域等涉及强相关的现象时,展现出了超越所有分析类密度泛函的出色行为,达到了甚至超越传统天梯顶端的精度。
然而,截至2025年对其实际算力工程应用的独立基准测试表明,深度神经网络泛函距离无缝应用仍有距离。俄罗斯Skoltech的研究团队在PySCF框架下针对DM21执行几何优化基准测试时发现,由于神经网络架构本身的内禀特性,其输出的交换相关能量和局部势能函数在空间坐标域上并不具备解析泛函那样的平滑特征。这种被放大的“非平滑性”数值噪声直接污染了基于Hellmann-Feynman定理的解析核梯度计算。为了获得足够平滑的梯度向量从而防止几何优化流程过早发散,研究人员被迫采用步长极小(0.0001至0.001 Å区间)的昂贵数值差分操作。这极大地拉长了分子结构弛豫和声子计算所需的单点执行周期,从而在很大程度上抵消了其预测能力上的优势。
6.2 跨越精度壁垒的终极探索:Microsoft Skala 泛函
为了在确保高度数值稳定性的同时一并解决高计算复杂度的难题,微软人工智能与科学研究院(MSR AI for Science)于2025年末发布了全新的深度学习泛函 Skala。Skala展示了当前AI与第一性原理物理结合的极致潜力,它的设计理念与物理机制包含以下突破:
放弃昂贵特征,回归数据驱动表示:有别于传统泛函依赖愈加复杂的、人为构造的高阶密度导数项以捕捉物理特征的思路,Skala仅接受相对廉价的元广义梯度近似特征(包括局域密度、密度梯度以及动能密度)作为模型输入。它利用神经网络直接将电子密度的不规则三维点云映射为包含复杂非局域信息的抽象标量空间,由算法自主挖掘其中的复杂相互作用函数。
极致紧凑的网络架构设计与标度保留:Skala模型包含一个参数量为 265,473 的局域分支以及一个极其轻量级的、通过跨网格点通信学习非局域相关性的非局域架构,总计参数量定格在仅仅 276,001 个。这种紧凑型架构不仅全面支持GPU加速执行,而且在理论上成功保持了与最基础的半局域DFT(LDA/GGA)完全一致的
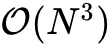 渐进计算复杂度。
渐进计算复杂度。通过 MSR-ACC 训练集逼近CCSD(T)极限:模型的预测效能归功于一项名为 MSR-ACC 的专有高质量数据集的支持。该数据集包含了多达8万个原子化能(TAE)标定数据,规模比之前所有的传统数据集大两个数量级,且其训练数据全部采用能达到 1 kcal/mol 绝对化学精度的波函数理论(如耦合簇单双重和微扰三重展开)生成。在广泛的主族热力学与动力学评测中,Skala在计算速度与低端半局域泛函并驾齐驱的条件下,达到了需要消耗天量资源的双杂化泛函(甚至高度参数化的波函数方法)的精度上限。
物理定律的涌现(Emergence of Constraints):最令理论学界关注的是,尽管Skala的神经网络架构在初始设计阶段去除了人为施加的大部分物理约束,但随着模型在极其广泛且多样的化学构型数据上完成迭代,符合真实物理本质的结构法则(例如遵循严格的动能相关分量演变要求)会自动从网络的隐空间中涌现出来。这种“物理信息启发”的机器学习工具(如开发中的 xcquinox 框架同样证明了这一点)宣告着纯粹解析的密度泛函开发范式正在落幕,取而代之的将是数据与神经网络交织重组隐式电磁拓扑的新时代。
七、 超越原子轨道的限制:无轨道密度泛函理论(OF-DFT)的复兴与极端条件探测
如果将研究的视野从百千万个原子构成的纳米团簇延展至包含数百万乃至上千万原子的介观相变系统,即便采用线性标度的Kohn-Sham DFT也由于依然需要构建巨大的辅助电子轨道集而耗尽显存。在这一背景下,回归Hohenberg与Kohn的原始初衷——直接丢弃任何形态的单电子波函数、完全以空间三维电子密度 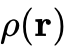 为唯一基础变量求解全部系统能量方程的无轨道密度泛函理论(Orbital-Free DFT, OF-DFT)——成为了终极解决方案。
为唯一基础变量求解全部系统能量方程的无轨道密度泛函理论(Orbital-Free DFT, OF-DFT)——成为了终极解决方案。
阻碍OF-DFT成为主流通用计算框架的长期瓶颈在于,科学界一直未能找到一种既不依赖波函数、又能以高度精确的方式计算体系总体动能分布的解析泛函(即动能泛函,Kinetic Energy Functionals, KEFs)。此外,由于失去了全轨道壳层屏蔽效应的描述,无轨道模拟必须配合专属设计的极高精度局部赝势(Local Pseudopotentials),极大制约了其对重元素体系的应用范围。
机器学习动能泛函与温稠密物质(WDM)的突破应用
步入2025年至2026年,机器学习算法直接介入到了动能密度映射之中,催生了OF-DFT领域的关键突破。
高精度泛函回归与网络拓扑:利用符号回归及各类高度特化的神经网络,理论学家终于构建出了计算精度能够逼近毫哈特里(mHa)量级的动能泛函。部分基于Matern52内核的模型不仅获得了比传统半局域动能泛函更高的精度,而且通过精巧的损失函数设计彻底消除了早先在预测动能势场时产生的剧烈空间振荡问题。进一步地,融合旋转等变图神经网络特征的模型(如 STRUCTURES25),被成功应用于包含大量复杂有机小分子的 QM9 数据库。该模型不仅无需经过复杂的自洽Kohn-Sham波函数收敛过程,还能迭代演化出具有明确物理意义的基态电子密度和处于严格化学精度范畴内的基态能量。
极端物质状态诊断的核心利器:OF-DFT极其低微且完美的线性计算资源标度,使其在一种最极端的物理环境——温稠密物质(Warm Dense Matter, WDM)研究中不可或缺。温稠密状态广泛存在于恒星内部(如白矮星和巨行星内核),并伴随着极高的压力和几千甚至几万开尔文的高温,是惯性约束核聚变(ICF)实验中的关键等离子体演化中间态。在如此高的热激发能量下,处于费米-狄拉克分布尾部的Kohn-Sham系统将不得不引入海量的高能虚拟空轨道(数量多达数千倍于基态),导致基于对角化程序的常规DFT软件因维度爆炸而瞬间崩溃。得益于纯密度的标量特性,OF-DFT可以极其平滑地扩展至有限温度泛函形式,辅以最新发布的完全热力学 meta-GGA 交换相关自由能演算法(如 fTSCAN 结构模型),使其能够毫无阻碍地捕捉这种等离子体中的量子电子气畸变行为。近期的大型国家实验室测试中,OF-DFT成功支撑了对极端液态碳及强磁场超热效应的X射线自由电子激光散射数据的底层理论解析。
八、 多尺度架构的终极重构:量子计算时代的混合电子结构演进
无论我们如何利用机器学习去弥补泛函设计中的经验性误差,抑或是利用超级计算机的海量算力展开矩阵,只要系统的核心基石仍然建立在密度平均场或单参考行列式的基础之上,它就无法在原理层面严格解决涉及强关联与高度多组态纠缠的量子核心难题(例如:催化反应循环中具有多重近并性能级的活性过渡金属簇、光系统复合蛋白的激发态辐射非绝热通道)。在这个层面上,真正具备模拟量子特性的技术体系必然走向硬件架构的最底层重构——量子计算。
即便在容错量子计算机(FTQC)能够处理全面多体问题的遥远未来到来之前,2025至2026年间的近期量子基准测试已经确立了一条极为明晰的发展路线:将经典高通量DFT代码作为低相关环境场引擎,与用于求解强相关组态的量子处理单元(QPU)构成的量子-经典混合多物理场工作流。
8.1 活性空间提取与投影嵌入机制
当前,针对规模超越100个量子比特以上的超大工业或生物化学体系计算已经逐步脱离粗糙的气相模型。通过系统性地部署投影嵌入机制(Projection-based embedding),庞大分子体系被物理拆解:计算成本较低的宏观蛋白质骨架、大块金属氧化物载体结构以及溶剂化环境,由传统的周期性DFT或经典分子力学算法予以解析,用于计算非活性的静电场并投射到核心区域;而包含强多参考特征的反应中心(即所谓活性空间,Active Space,如罕见疾病代谢抑制剂形成共价键的中心或者金属表面游离吸附中间体)的复杂相关能量,则交由量子态层析网络进行运算。通过预先执行局域化分子轨道的特定优化旋转方案,量子哈密顿量的 -范数(评估量子线路资源与采样开销的核心指标)被成功缩小,极大缓解了带噪中型量子(NISQ)计算的运行时长。
8.2 新型量子算法框架与表面催化基准测试
为了替换经典算法在处理完全活性空间组态相互作用(CASCI)时的指数爆炸阻碍,变分量子本征求解器(VQE)已经进一步演化出诸多适应性方案。目前广受重视的架构包括分解幺正耦合簇(fUCC)、针对特定分子拓扑生成自适应 ansatz 算符的 ADAPT 算法,以及旨在跨过多参考计算阻碍的量子选择配置相互作用(QSCI)。
一项标志性的多相催化基准测试(以探究銠掺杂金红石二氧化钛表面的 NO 解离与吸附为例,Rh@TiO2(110) 体系)展示了这些量子拟设的极限能力。在该模型中,处于表面解离极限状态的游离 NO* 自由基与具有配对单电子的 Rh(IV) 活性中心会形成复杂的纠缠多体共振态,这是纯经典半局域DFT体系极难模拟定量的静态误差地带。通过将状态平均的 CASSCF 参考轨道无缝导入量子处理器,不仅获得了对这种涉及多组态断键和成键动力学的全覆盖映射,还为未来评估光刻机极端紫外光源的非绝热动力学和新型储能材料在充放电循环下的共振非弹性X射线散射(RIXS)铺平了道路。
九、 展望
过去的一个世纪见证了人类探索原子乃至电子波函数极限的物理学历程。密度泛函理论的演进轨迹——从Thomas-Fermi的纯粹统计猜想,历经Hohenberg与Kohn的数学奠基和KS参考系统的建立,在“雅各布天梯”上不断通过经验与解析梯度补偿逼近实验值的攀登——其本质是对“物理表述精度、多体相互作用的复杂度与可用算力资源边界”之间永恒权衡的探索。
尽管经典DFT方法学由于衍生出无数高度分化的计算包和数千万条海量物性数据而促生了整个计算材料科学,但在自相互作用、半导体带隙预测偏离以及强静态相关灾难这三大底层内禀物理缺陷面前,基于传统解析微积分的单平均场架构已不可挽回地步入其能力的极限区间。
2025年后,我们正处于理论体系发生根本性范式交叠的前夜。通过放弃人工干预物理特征并引入诸如 Skala 和旋转等变图网络的纯数据驱动架构,深度学习不仅彻底解开了高阶物理响应精度与计算渐进标度间的死结,并成功实现了物理内秉约束机制在网络隐空间中的自主涌现。与此同时,神经网络模型引导下的高精度动能泛函更是打破了数十年无轨道模型(OF-DFT)的沉寂,让宏观热等离子体及极端物质状态下的百千万原子体系计算成为常规。最终,在新兴的异构量子-经典体系计算管线中,DFT的职能正在完成蜕变:它非但不会衰退,反而将演进为全尺度量子力学探测中最为强悍、可靠的前置环境算力底座,专门处理巨量且连续的弱关联均场空间;而将自然界最隐秘的、最深邃的复杂多体纠缠行为,毫无保留地释放回指数级维度的量子比特矩阵。这一跨越尺度、跨越架构的物理算法融合,必将引领未来物质科学进入预测的绝对自由王国。