


出处:Coronado M, Fajardo G, Nguyen K, Zhao M, Kooiker K, Jung G, Hu DQ, Reddy S, Sandoval E, Stotland A, Gottlieb RA, Bernstein D. Physiological Mitochondrial Fragmentation Is a Normal Cardiac Adaptation to Increased Energy Demand. Circ Res. 2018 Jan 19;122(2):282-295.
作者介绍:

Abstract
摘要
Introduction
前言
心肌由于节律性收缩对ATP具有极高需求,因此对能量稳态的改变尤为敏感¹。心脏最初通过利用局部ATP储备和无氧代谢来应对需求增加,而持续性需求增加则通过增强线粒体氧化磷酸化以及线粒体动力学(分裂、融合、线粒体自噬和生物发生)的改变来维持有氧ATP生成²˒³。在心脏中,线粒体分裂及其后续碎片化通常由缺血等病理性应激引起,并被视为线粒体功能障碍的标志⁴˒⁵。然而,近期研究表明,抑制线粒体分裂同样会导致心脏功能受损⁶⁻⁹,这提示线粒体分裂对于维持正常线粒体稳态同样具有重要意义。Song等表明,条件性Drp1敲除(−/−)小鼠虽然保留了线粒体功能,但线粒体自噬增加,并最终发生心力衰竭¹⁰。其他研究则表明,线粒体分裂受损会导致功能异常线粒体的积聚¹¹。相反,Drp-1抑制剂mdivi-1可减少线粒体自噬,并挽救压力超负荷诱导的心力衰竭¹²。还有研究提出,在心肌细胞中,线粒体分裂并不会显著改变线粒体形态,而主要是为了通过不对称分裂和线粒体自噬促进功能异常线粒体的清除所必需的⁸˒¹⁰。由于上述研究均依赖于对Drp1等动力学调控因子的转基因操作,或是在缺血等病理状态下开展,因此,线粒体分裂作为正常心脏生理适应机制的作用仍不清楚。
Methods
研究方法
01
数据与材料可获得性
微阵列数据已按MIAME格式提交至NCBI基因表达综合数据库(Gene Expression Omnibus,GEO)。数据请求可联系:Daniel Bernstein博士,750 Welch Road Suite 325, Palo Alto, CA 94304。danb@stanford.edu。有关实验方法的详细描述,请参见在线数据补充材料中的材料与方法部分。
02
动物实验方案
8–10周龄雄性FVB/NJ小鼠购自Jackson Laboratory(Bar Harbor,ME)。小鼠在配备同步测定VO2和VCO2功能的跑台上进行运动(Columbus Instruments,Columbus,OH),并按照我们既定的实验方案测定跑步距离和呼吸交换率(RER)¹⁹。小鼠采用两种不同的运动方案。第一种为亚极量运动:以20 m/min的速度、5°恒定坡度跑1 h。采用该方案评估运动能力时,使小鼠以20 m/min的速度、5°恒定坡度跑至力竭,并记录总跑步距离。第二种方案用于评估最大运动能力:坡度每3 min增加2°,速度每3 min增加2 m/min。运动时间定义为从开始运动至力竭,或在电击网格上静止10 s的时间。运动距离按总垂直移动距离(m)计算,即:跑台速度(m/min)×坡度百分比(Sin θ)×运动时间(min)。做功(J)按体重(kg)×总垂直移动距离(m)×9.8计算。
03
β-肾上腺素能信号及线粒体动力学调节
为评估亚型特异性的β-肾上腺素能受体(β-AR)信号,我们使用了由本实验室在FVB遗传背景上构建的β2-AR敲除(−/−)小鼠¹⁹。为评估线粒体分裂,小鼠腹腔注射Drp1-Fis1抑制剂P110(0.5 mg/kg,由Daria Mochly-Rosen博士提供)或Mdivi-1(25 mg/kg),随后立即进行运动测试。对照处理分别采用生理盐水、TAT47–57肽(0.35 mg/kg)或生理盐水中2% DMSO。为评估β1-AR信号的作用,小鼠于最大运动挑战前90 min腹腔注射β1-AR拮抗剂美托洛尔(5 mg/kg)。为评估心脏特异性的线粒体分裂,Drp1loxp/loxp C57B/6NJ小鼠由Katsuyoshi Mihara博士和Masatoshi Nomura博士提供,并与my
h6-MER-Cre-Mer C57B/6NJ小鼠品系(Jackson Laboratory,Bar Harbor,ME)杂交;随后通过连续5 d腹腔注射他莫昔芬(20 mg/kg)诱导基因重组。第7天进行实验。接受他莫昔芬处理的同龄雄性Drp1+/+Cre+/+和Drp −/−Cre−/−小鼠作为对照。
04
异丙肾上腺素、多柔比星及抑制剂给药
HL-1细胞和心肌细胞给予10 μM异丙肾上腺素(ISO)处理1 h。HL-1细胞在ISO暴露前30 min预处理300 nM亚型特异性抑制剂CGP 12177(β1拮抗剂)、ICI 118551(β2拮抗剂),以及1 μM L748,337(β3拮抗剂)和普萘洛尔(β1和β2拮抗剂)。细胞还在ISO暴露前30 min预处理30 μM线粒体分裂抑制剂Mdivi-1。
05
统计学分析
数据以均数±均数标准误表示。符合正态分布的两组比较采用Student t检验。符合正态分布的多组比较采用方差分析(ANOVA)并进行多重比较分析(Tukeys)。两组间非参数数据比较采用Mann-Whitney U检验。微阵列数据的统计分析采用Stanford microarray database software、Significance Analysis of Microarrays(SAM)、Database for Annotation, Visualization, and Integrated Discovery、Hi-Throughput GOMiner及TIGR TM4软件进行。多重比较采用错误发现率(FDR)分析进行校正,并采用Fisher精确检验,以p<0.01作为基因差异显著改变的判定标准。为控制多重比较,我们将错误发现率控制在5%。微阵列数据已按MIAME格式提交至NCBI基因表达综合数据库(GEO)。
Results
研究结果
01
运动诱导“生理性”线粒体碎片化
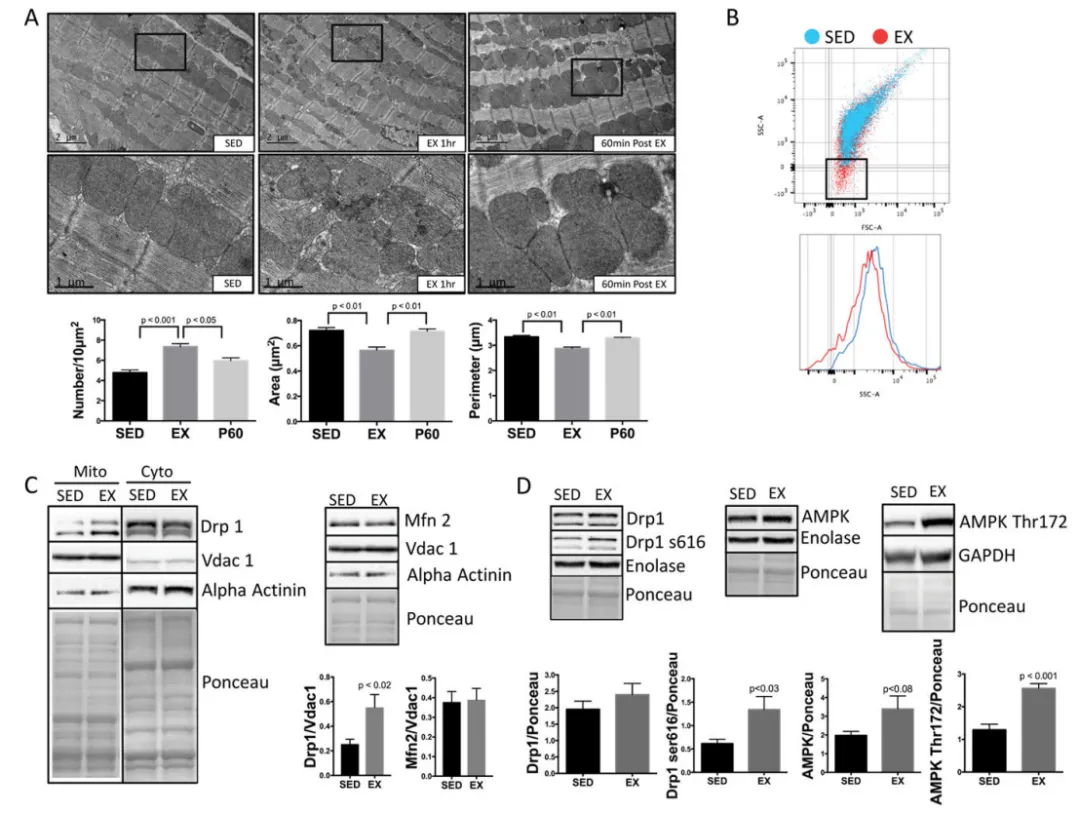
在接受1 h亚极量跑台运动的小鼠中¹⁹,定量电子显微镜结果显示心脏线粒体发生了显著碎片化:数量增加,面积和周长减小(图1A)。我们进一步采用流式细胞术侧向散射面积(SSA)验证了运动诱导的线粒体碎片化,结果表明,与缺乏运动对照组相比,运动小鼠的线粒体体积更小(图1B)。SSA结果通过已知粒径微球(2 μm、1 μm、0.5 μm、0.2 μm)校准得到验证(在线图I)。线粒体分裂可通过线粒体分裂介导因子Drp1从胞质转位至线粒体组分来证实²²。我们通过胞质蛋白alpha-actinin丰度较低以及线粒体Vdac1丰度较高来评估线粒体组分的纯度。运动诱导促分裂介导因子Drp1转位至线粒体(图1C),并诱导分裂激活位点Drp1丝氨酸616发生磷酸化(图1D)。促分裂的AMP活化蛋白激酶(AMPK)也被激活,表现为其激活位点苏氨酸172磷酸化增加(图1D);AMPK总蛋白表达无变化(虽有升高趋势,但未达到统计学显著性)。相反,促融合介导因子Mfn2无变化(图1C)。运动恢复1 h后,线粒体结构开始恢复至缺乏运动时的形态(图1A)。
02
生理性线粒体碎片化是获得最大运动表现所必需的
为确定生理性碎片化对于实现最大运动表现是否必需,我们给予小鼠P110处理¹⁹。P110通过阻断Drp1与Fis1相互作用、抑制Drp1向线粒体转位,从而抑制线粒体分裂(图3A)³⁸。随后,小鼠接受亚极量和最大运动能力测试。P110处理使亚极量运动时间下降45%,亚极量运动距离下降45%(图3B)。为尽可能控制体重差异带来的影响,我们计算了总做功(J),结果发现P110处理小鼠的亚极量运动做功下降38%。同样,在最大运动能力测试中,P110处理小鼠的最大运动时间下降16%,运动距离下降37%,总做功下降38%(图3C)。P110处理小鼠也较载体处理小鼠更早达到无氧阈值(RER>1.0),提示在运动过程中阻断线粒体分裂会更早启动不依赖氧化磷酸化的(无氧)ATP生成。在运动早期(前5–10 min),P110对呼吸交换率(RER)无影响(在线图IV),这表明生理性碎片化的重要性仅在能量需求持续增加较长时间后才显现。亚极量和最大运动能力测试均可诱导线粒体碎片化。P110处理可完全阻断亚极量运动诱导的碎片化,而对最大运动诱导的碎片化仅部分阻断(图3B、3C),这进一步表明生理性线粒体分裂对于达到最大运动能力是必需的。由于线粒体分裂还可通过Fis-1非依赖机制发生³⁹,我们进一步采用Drp1 GTP酶抑制剂Mdivi-1验证Drp-1在生理性碎片化中的作用。与P110类似,Mdivi-1处理小鼠的最大运动能力下降(在线图V)。
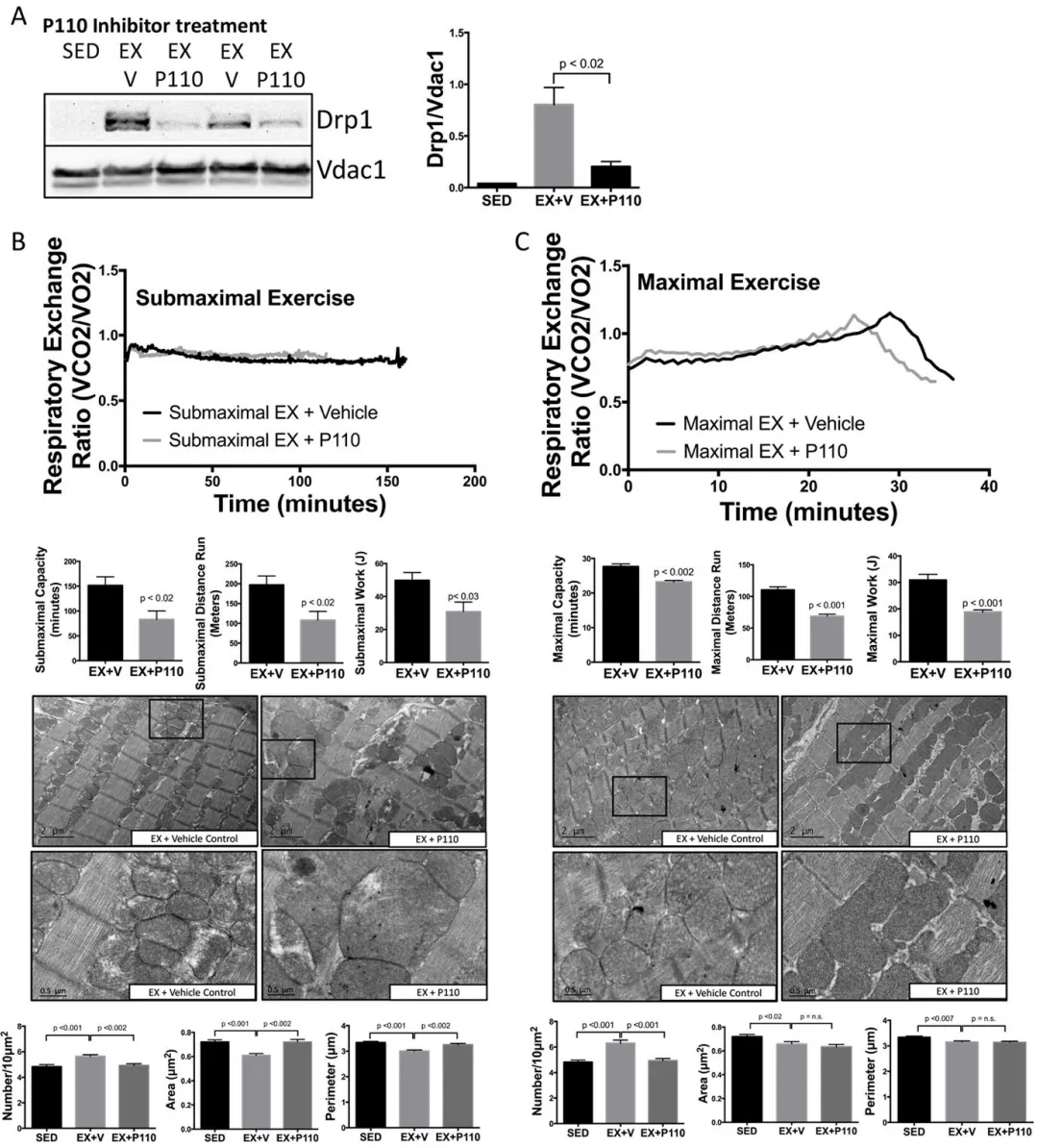
图3. 线粒体碎片化是亚极量和最大运动能力所必需的
(A)Drp1抑制剂P110(0.5 mg/kg,腹腔注射)可降低Drp1向线粒体的转位,与TAT载体(V)对照相比明显减少(每组n=3)。
(B)阻断Drp1可降低亚极量运动,以及(C)最大运动的运动时间(min)、运动距离(m)和做功(J)。P110处理还可阻止线粒体碎片化,其证据是在(B)亚极量运动和(C)最大运动过程中,线粒体数量均未出现增加(每组n=5)。
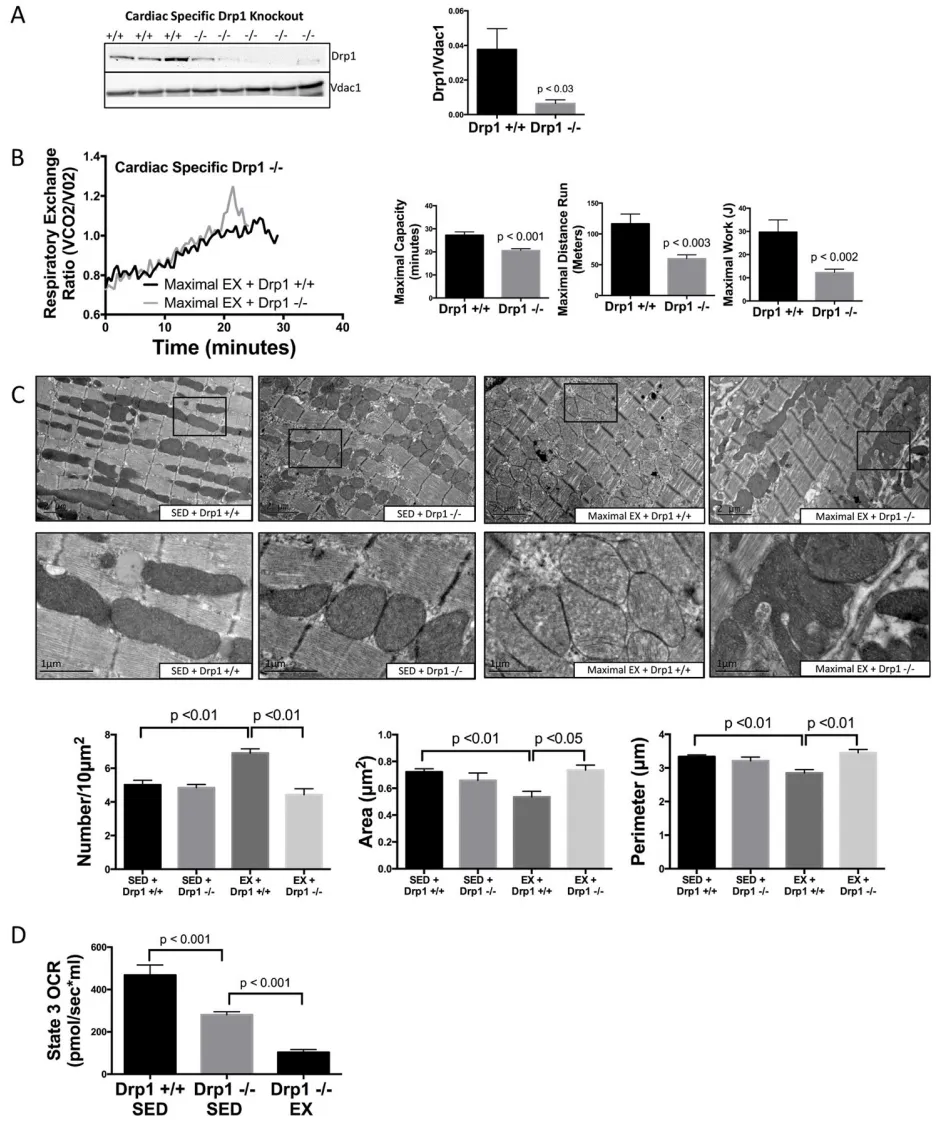
图4. 心脏线粒体碎片化是达到最大运动能力所必需的
(A)通过将Drp1loxp/loxp小鼠与myh6-MER-Cre-Mer小鼠杂交,并连续5 d给予他莫昔芬处理(20 mg/kg,腹腔注射),构建心脏特异性条件性Drp1 −/−小鼠。
(B)Drp1 −/−使最大运动时间(min)、运动距离(m)和做功(J)降低;并且(C)阻止了运动诱导的线粒体碎片化,但产生了形态极其异常的线粒体,包括细长型(箭头所示)或高度分节型(下图)。静息状态下Drp1 +/+与Drp1 −/−之间未观察到线粒体形态变化(每组n=3–5个生物学重复,每个生物学重复至少取10个视野后求平均)。
(D)采用Oroboros oxygraph测定并按50 μg线粒体归一化后发现,静息和运动状态下的Drp1 −/−小鼠均表现出线粒体功能降低(每组n=5)。
03
β1-AR信号诱导生理性线粒体碎片化并增强线粒体功能
交感神经系统是满足运动血流动力学需求的主要机制,它通过增加心率和心肌收缩力来提高能量需求。这一反应由β-肾上腺素能受体(β-AR)信号介导⁴⁰,主要通过蛋白激酶A(PKA)介导细胞内钙浓度([Ca2+]i)升高,而钙离子既调控线粒体ATP生成,也调控线粒体动力学⁴¹。为考察β-AR信号在线粒体碎片化中的作用,我们用MitoTracker标记HL-1心肌细胞,并给予生理剂量(10 μM)的β-AR激动剂异丙肾上腺素(ISO)处理1 h,结果导致线粒体碎片化增加(图5)。β1-AR拮抗剂CGP12177(300 nM)可阻断ISO诱导的碎片化,而β2-AR拮抗剂ICI118,551(300 nM)则不能,表明线粒体动力学受β1-AR亚型特异性调控。
图5. β1-AR信号在HL-1细胞中诱导线粒体碎片化
细胞给予ISO(10 μM)处理1 h后,采用共聚焦显微镜成像。ISO可增加线粒体碎片化(表现为周长和面积减小、数量增加),且这一效应可被CGP12177(β1拮抗剂)阻断,但不能被ICI118551(β2拮抗剂)阻断(每组n=13–25个细胞)。细胞定量采用Image J自定义宏程序完成。
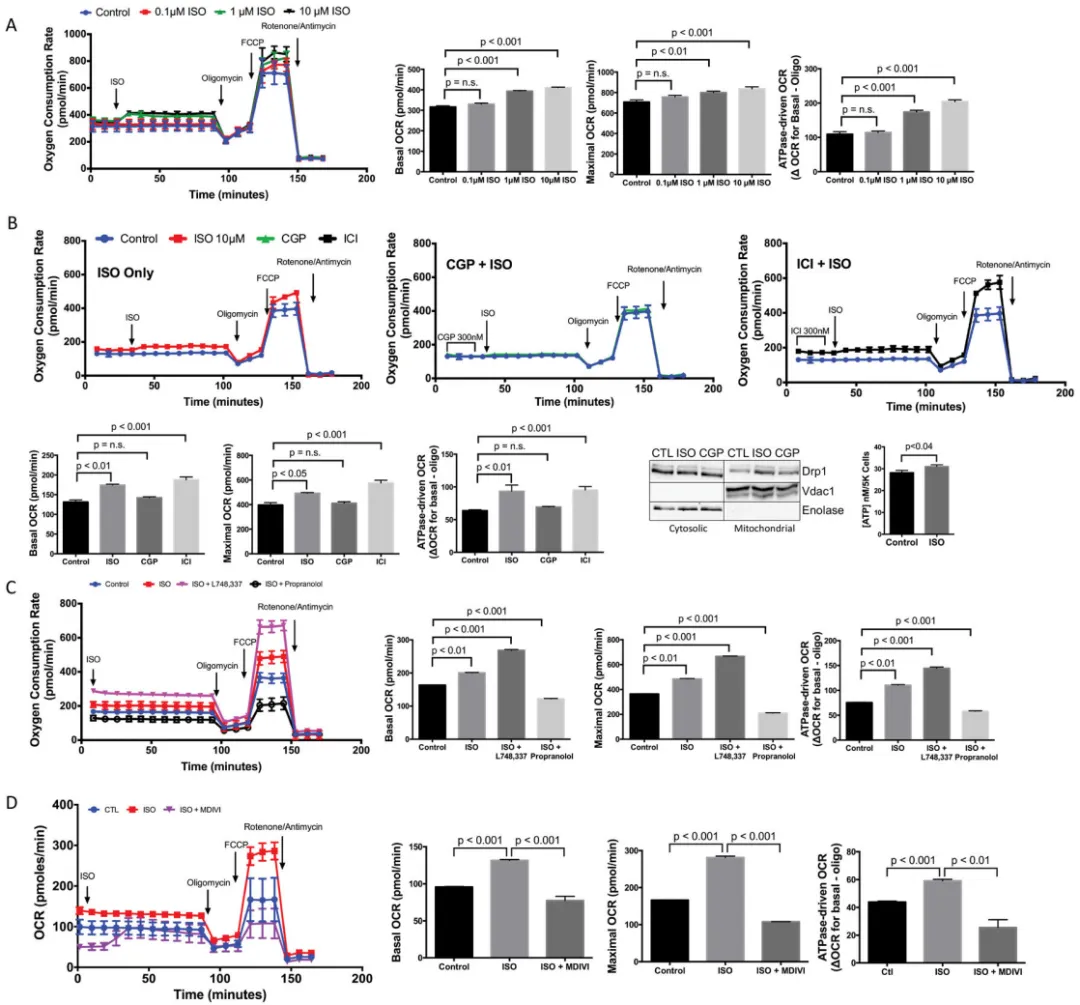
图6. β1-AR信号增强HL-1细胞线粒体呼吸,而这一作用受到β2-AR和β3-AR信号的拮抗
(A)ISO自1 μM起即可剂量依赖性地增加基础呼吸、最大呼吸及ATP酶驱动呼吸(每组n=5)。
(B)这种增加可被CGP阻断,并可被ICI进一步增强(每组n=4)。ISO可增加Drp1向线粒体转位,而这一效应可被CGP阻断。ISO还可显著增加ATP生成(每组n=7)。
(C)ISO增强的呼吸在联合β3AR拮抗剂L748,337时呈协同增加,而在普萘洛尔(β1/β2拮抗剂)处理时被阻断(每组n=5)。
(D)采用Mdivi-1抑制线粒体分裂可阻断ISO诱导的线粒体呼吸增强(每组n=5)。
04
体内删除β2-AR可导致Drp1介导的线粒体分裂并增强运动能力
为进一步提供β1-AR介导线粒体碎片化的体内证据,我们评估了β2-AR敲除小鼠(β2−/−)。该模型表现为慢性生理水平β1-AR信号增强,而β1受体密度或功能并未改变,也不存在β2-AR及β2-AR-Gi信号的拮抗效应²⁰˒⁴²。重要的是,我们此前已证实β2−/−小鼠具有正常的心脏结构、功能和生存率²⁰。与我们的体外结果一致,β2-AR−/−小鼠表现出明显的线粒体碎片化(图7A)、Drp1转位以及线粒体自噬调控因子PINK1下调(图7B)。在采用全基因组筛查鉴定出的35个上线或下调的线粒体相关基因中(在线表I),发生变化的唯一线粒体动力学调控因子是beclin-1,且这一线粒体自噬介导因子呈下调,提示线粒体动力学调控因子的转录水平调节并不是慢性β-AR诱导线粒体动力学变化的主要机制。值得注意的是,尽管β2−/−小鼠存在显著的线粒体碎片化,但其基础线粒体功能并未受损(图7C),且与WT相比,β2−/−小鼠的运动跑步时间、距离和做功均增加(图7D)。为进一步证实增强的β1-AR信号的作用,我们在运动挑战前90 min腹腔注射β1特异性拮抗剂美托洛尔(5 mg/kg)以阻断β1-AR信号。β2−/−小鼠增强的运动做功在β1-AR阻断后显著下降(图7D),这进一步证明线粒体分裂和碎片化在心脏运动适应中发挥重要作用。
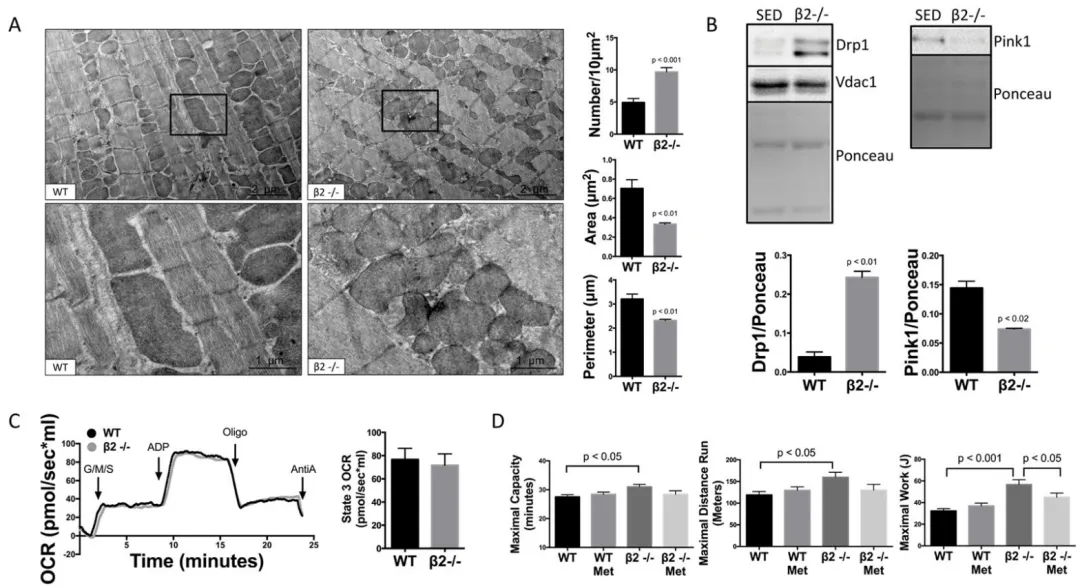
图7. β2-AR缺失可导致生理性线粒体分裂并增强运动能力
(A)β2−/−小鼠作为一种无β2-AR拮抗的生理水平β1-AR信号模型,与WT同窝小鼠相比,表现出显著的线粒
体碎片化,具体为数量增加以及面积和周长减小(每组n=3–5)。
(B)静息状态下,β2−/−小鼠中Drp1向线粒体转位增加,而PINK1减少(每组n=3)。
(C)尽管存在明显的线粒体碎片化,β2−/−小鼠按50 μg线粒体归一化后,经Oroboros oxygraph测定的基础呼吸仍保持正常(每组n=4)。
(D)β2−/−小鼠的
运动跑步时间、距离和做功均增强,而这种增强可被美托洛尔介导的β1-AR抑制所阻断(每组n=8)。
Discussion
讨论
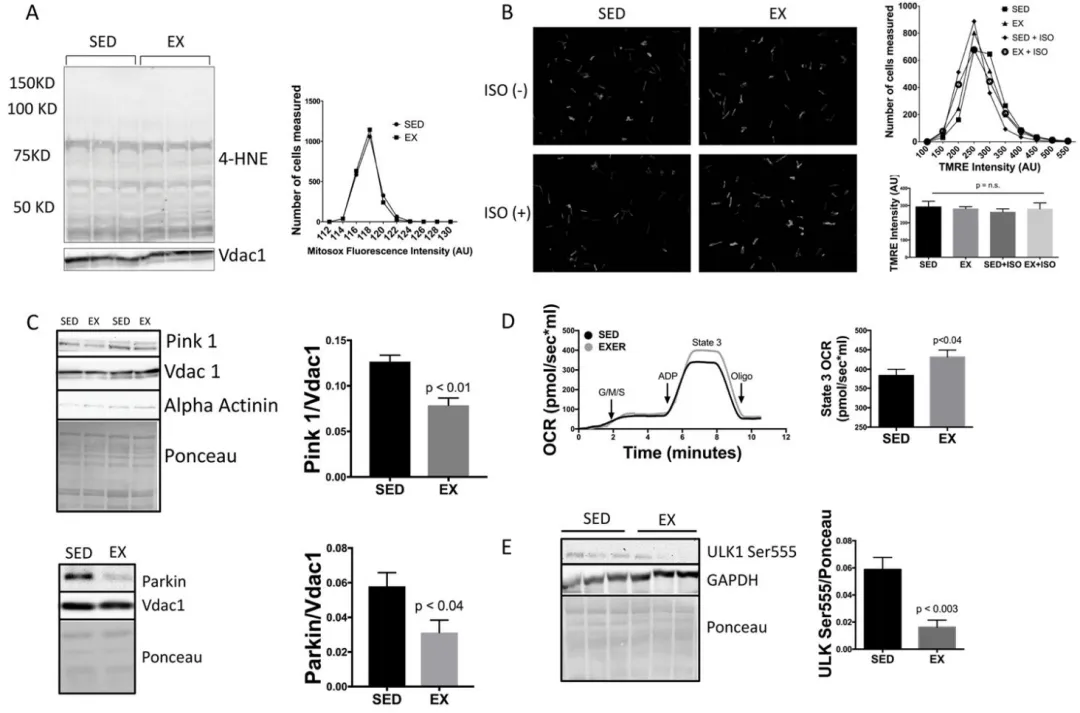
线粒体分裂传统上一直被认为与病理性应激及线粒体功能受损相关,然而,近年来的研究提示,线粒体分裂在线粒体正常心脏稳态调控中也发挥作用⁶˒⁸˒¹⁰。在本研究中,我们证明了心脏线粒体分裂和碎片化并不局限于病理状态,它也是正常生理性应激(如亚极量运动)期间线粒体功能正常生理调控的组成部分。这与近期关于分裂对维持正常线粒体质量控制必不可少的报道一致⁵。然而,由于这些研究依赖于基因操作,如Drp-1敲除,或是在病理背景下开展,因此,本研究首次证明了线粒体碎片化在正常心脏生理中的作用。作为线粒体能量代谢的正向介导因素,生理性碎片化对于达到最大运动表现至关重要。当采用Drp1抑制剂P110、Mdivi-1,或通过短期条件性心脏特异性Drp1敲除阻断线粒体分裂和碎片化时,运动能力显著下降,更早转向非依赖氧的ATP生成机制,这一点可由达到无氧阈值时间提前得到证实,同时ATP合酶依赖的状态3呼吸显著降低。Benard等也报道了线粒体分裂对生物能量学的类似影响,其研究显示,在HeLa细胞中沉默Drp1会降低状态3呼吸、ATP合成速率以及葡萄糖饥饿条件下的存活率⁴⁸。Drp1沉默所见线粒体功能下降,可能与其在线粒体重塑、嵴开放及膜间隙扩张中的作用有关⁴⁸˒⁴⁹。其他细胞系统中的既往研究支持这样一个概念,即分裂、融合和线粒体自噬是足够迅速的过程,能够在运动等生理性应激因素作用的时间范围内发生⁵⁰⁻⁵²。本研究的局限之一在于,目前尚无方法可直接在体内运动过程中测量线粒体功能,因此我们的测量结果受到从运动后小鼠心脏中分离线粒体所需时间延迟的影响。因此,我们观察到的线粒体功能轻度升高,可能低估了运动期间线粒体功能增强的程度。我们在此的关键发现是:我们观察到了线粒体分裂,但并未伴随功能下降。
关于缺血/再灌注损伤(氧供应受限)、多柔比星毒性(线粒体氧化应激)或糖尿病(代谢应激)等病理性应激因素下线粒体动力学反应,我们课题组及其他研究者均已有报道,结果均显示线粒体分裂和碎片化被激活、ROS生成增加,并通过线粒体自噬/自噬启动对功能异常线粒体的清除⁴˒²³˒²⁴˒³³˒³⁴。这与亚极量运动(代谢需求增加)等生理性应激形成对比:此时ROS并不增加,线粒体膜电位得以维持,从而阻止PINK1/Parkin的激活,并抑制线粒体自噬。有趣的是,尽管AMPK被激活⁵³,促自噬蛋白ULK-1在亚极量运动中却处于失活状态。ULK-1失活可能由运动期间被激活的磷酸酶介导,例如双特异性蛋白磷酸酶1,其可作用于ULK1⁵⁴,并在心脏中表达⁵⁵,且已被证明可在运动时被激活⁵⁶。
He等此前的一项研究表明,运动期间心脏自噬被激活³⁷。相比之下,我们的亚极量运动模型并未诱导线粒体自噬或自噬,这支持了这样一种观点:在无线粒体功能障碍的情况下,这些过程在亚极量运动期间是失活的。我们的模型与He等研究的一个差异在于:我们让小鼠以其最大能力的80%、低于无氧阈值的水平,以20 m/min恒速运动1 h,且未进行预训练;而他们的研究则是先让小鼠进行2 d运动训练(8–10 m/min,5–10 min,10°坡度),然后于第3天进行最大运动。其他研究已表明,运动训练期可诱导自噬⁵⁷。为检验本研究与He等在自噬方面差异是否由运动应激水平不同所致,我们让小鼠运动至最大能力,结果显示,这同样可诱导分裂,但Parkin并未增加(尽管与亚极量运动研究不同,Parkin也未下降),且仍无自噬证据(在线图IIIC)。因此,尽管运动过程中可能存在分级的线粒体分裂水平,即亚极量运动诱导无线粒体自噬/自噬参与的生理性碎片化以增强呼吸,而极限/最大运动诱导更高水平的分裂,但在我们的所有模型中,仍未观察到自噬被激活。Kruse等在接受亚极量运动(70% VO2max,1 h)患者的骨骼肌中也发现了类似结果:Drp1在运动后即刻被激活,而LC3下降。关键的是,LC3仅在运动后3 h恢复期才增加⁵⁸,这提示自噬可能是运动恢复期的一个特征。除此之外,还有若干差异可解释我们结果与He等不同。我们的亚极量运动研究是在FVB/NJ遗传背景小鼠中进行,而He等使用的是C57Bl/6背景;已有研究表明,与FVB/NJ相比,C57Bl/6的总体运动能力较低⁵⁹,因此相似的运动方案可能给C57Bl/6小鼠带来更大的应激。重要的是,我们还发现,与自噬相关的蛋白在亚极量运动恢复期迅速增加(最早可于5 min出现,数据未显示),因此,为评估运动期间真实的自噬状态,必须立即采集心脏组织。最后,我们不能排除急性亚极量运动期间自噬通量发生变化的可能性,因为在未合并缺血或饥饿等更强附加应激的条件下,我们未能在氯喹处理后观察到自噬标志物增加。尽管存在这一局限,我们仍未见线粒体自噬被激活的证据,因为急性运动时PINK1和Parkin水平均下降,更重要的是,线粒体膜电位和功能得以维持。
我们进一步表明,生理性碎片化部分由β1-AR信号增强所介导,其可激活Drp-1并增加线粒体呼吸。我们通过联合使用Mdivi-1和ISO处理细胞加以证实,结果显示ISO刺激的线粒体呼吸被阻断。这些数据进一步证明,生理性线粒体碎片化与线粒体功能增强而非功能恶化相关。有趣的是,应用β2-AR拮抗剂ICI或β3-AR拮抗剂L748,337后,线粒体呼吸增加幅度均高于单独使用ISO。ICI同时阻断β2-AR-Gs和β2-AR-Gi信号,因此增强了β1-AR-Gs信号⁶⁰。β3-AR同样已被证明可偶联Gi⁴⁷。因此,使用ISO + ICI或L748,337时,β1-AR受到更强刺激,从而导致较单独ISO更高的呼吸增强。最后,已知β3-AR信号可在棕色脂肪组织中激活线粒体融合通路⁴³,这可能会对抗心脏中β1-AR诱导的线粒体分裂。然而,β3-AR在小鼠⁶¹和人⁶²心室肌细胞中的表达水平均很低。因此,与其他组织相比,β3-AR信号在运动中心脏中的作用可能没有那么重要。
另一种无病理背景下的碎片化模型是β2-AR敲除,在这种情况下β1-AR信号本身并未增加,但失去了β2-AR(包括其抑制性Gi信号)的拮抗作用⁴²。这些小鼠表现出明显的线粒体碎片化,但无线粒体功能受损证据,心脏结构和功能正常,且运动能力增强;而这种增强可被β1-AR抑制所阻断,从而证实了我们体外研究中β1-AR亚型特异性调控线粒体动力学的发现,并进一步证明了生理性碎片化过程的存在。本研究这部分的一个局限在于,在体内无法选择性阻断β1-AR刺激对线粒体分裂的作用,而不影响β1-AR刺激对心脏功能的整体效应。因此,我们在体外清晰显示的β1-AR介导分裂增加,可能只是体内介导该过程的诸多通路之一,例如,WT小鼠总运动能力不受美托洛尔影响(图8D),以及我们此前已证明β1-AR −/−小鼠具有正常运动能力⁶³,均支持这一点。其他可能参与运动诱导分裂的通路还包括β3-AR和AMPK信号通路。然而,在β2-AR −/−背景下,在线粒体分裂和碎片化静息状态即已增加的条件中,β1-AR受体在介导线粒体分裂中的重要作用得到了证实。
线粒体碎片化既可视为一种主动过程(单个线粒体的切割),也可视为一种形态学结果(线粒体数量增多且体积缩小),而两者并不一定等同。运动在Drp1−/−小鼠中诱导出的奇特线粒体形态(图4C),我们称之为“拼图样线粒体”,提示运动正在主动刺激线粒体分裂,但该过程在线粒体真正分裂前被中断。这进一步表明,Drp1等介导因子具有更复杂的作用。
Conclusion
研究结论