



HTqPCR:R 中实时定量 PCR 数据高通量分析与可视化
软件功能
HTqPCR 是为 R 统计计算环境 (www.r-project.org) 开发的,将在所有主要平台上运行,并作为开源提供。Core R 和 Bioconductor 包是唯一的软件依赖项,该包包含详细的教程。
1 数据输入要求
输入数据格式由制表符分隔的文本文件组成,其中包含 Ct 值、特征标识符(基因、microRNA 等)和其他(可选)信息。数据文件可以是用户格式的纯文本,也可以是序列检测系统 (SDS) 软件的直接输出。在内部,此信息体现为 qPCRset 类的实例,类似于通常用于表示微阵列分析中荧光数据的 ExpressionSet 对象。
2 可视化功能
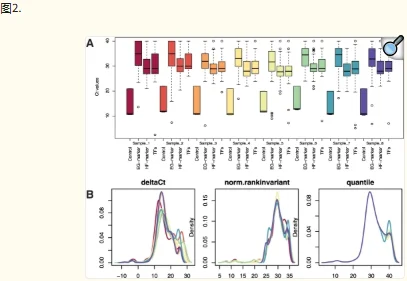
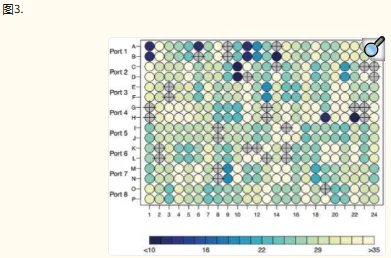
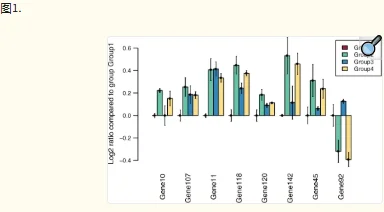
HTqPCR 包含多种数据可视化功能。一个或多个样本中的基因子集可以用条形图表示,显示与校准品样本相比的绝对 Ct 值或倍数变化(图 1)。可以通过密度分布、箱线图、散点图和直方图等诊断辅助工具来评估样本的数据质量控制,其中一些可以根据特征的各种属性进行分层(图 2)。当在多孔板或其他空间定义的布局中进行qPCR测定时,可以相应地绘制Ct值以可视化任何空间伪影,例如边缘效应(图3)。可以使用主成分分析、热图或树状图对样本或基因进行聚类。
3 Ct质量控制
单个 Ct 值是 qPCR 结果不确定性的主要来源。这可能是由于扩增条件的固有偏差(可变引物退火、扩增子序列含量、次优反应温度或盐浓度等)引起的,或者当初始模板浓度不足以产生超过最小检测阈值的拷贝数时。在 HTqPCR 中,Ct 值的可靠性可以单独或跨重复评估。通过用户可调整的参数,所有值都被标记为“正常”、“未确定”或“不可靠”之一,并且这些信息在整个分析过程中传播。非特异性过滤可用于去除在样本中标记为“未确定”和/或“不可靠”的基因,或在归一化后具有低变异性(即未差异表达)的基因。
#GBD数据库 #Meta #charls数据库 #sci #Chun数据库 #生信分析 #医学sci #机械学习 #孟德尔随机化 #UKB数据库
软件功能
HTqPCR 是为 R 统计计算环境 (www.r-project.org) 开发的,将在所有主要平台上运行,并作为开源提供。Core R 和 Bioconductor 包是唯一的软件依赖项,该包包含详细的教程。
1 数据输入要求
输入数据格式由制表符分隔的文本文件组成,其中包含 Ct 值、特征标识符(基因、microRNA 等)和其他(可选)信息。数据文件可以是用户格式的纯文本,也可以是序列检测系统 (SDS) 软件的直接输出。在内部,此信息体现为 qPCRset 类的实例,类似于通常用于表示微阵列分析中荧光数据的 ExpressionSet 对象。
2 可视化功能
HTqPCR 包含多种数据可视化功能。一个或多个样本中的基因子集可以用条形图表示,显示与校准品样本相比的绝对 Ct 值或倍数变化(图 1)。可以通过密度分布、箱线图、散点图和直方图等诊断辅助工具来评估样本的数据质量控制,其中一些可以根据特征的各种属性进行分层(图 2)。当在多孔板或其他空间定义的布局中进行qPCR测定时,可以相应地绘制Ct值以可视化任何空间伪影,例如边缘效应(图3)。可以使用主成分分析、热图或树状图对样本或基因进行聚类。
3 Ct质量控制
单个 Ct 值是 qPCR 结果不确定性的主要来源。这可能是由于扩增条件的固有偏差(可变引物退火、扩增子序列含量、次优反应温度或盐浓度等)引起的,或者当初始模板浓度不足以产生超过最小检测阈值的拷贝数时。在 HTqPCR 中,Ct 值的可靠性可以单独或跨重复评估。通过用户可调整的参数,所有值都被标记为“正常”、“未确定”或“不可靠”之一,并且这些信息在整个分析过程中传播。非特异性过滤可用于去除在样本中标记为“未确定”和/或“不可靠”的基因,或在归一化后具有低变异性(即未差异表达)的基因。
#GBD数据库 #Meta #charls数据库 #sci #Chun数据库 #生信分析 #医学sci #机械学习 #孟德尔随机化 #UKB数据库