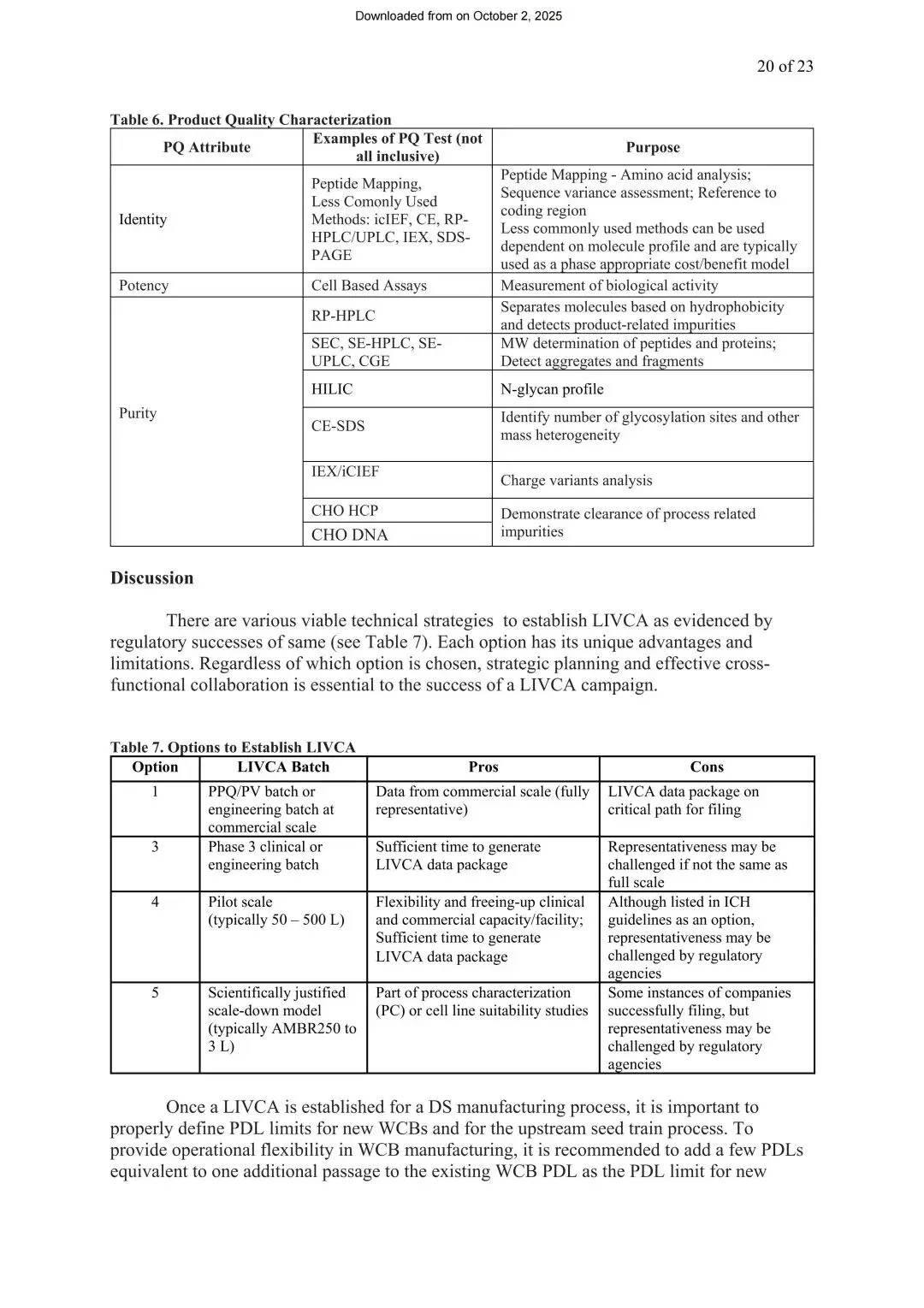
点击蓝字 关注我们
生物药的生产,本质上是让细胞当“活体工厂”。以最主流的抗体药为例,CHO细胞被植入目的基因后,要在生物反应器里不断分裂增殖,源源不断地产出药物蛋白。从最初冻存的主细胞库(MCB),到最终几千升规模的商业化生产,细胞中间已经经历了大量次分裂。
分裂次数越多,风险也在累积:基因可能发生突变,潜伏在细胞里的病毒可能被激活,蛋白质的产量和质量也可能随之漂移。正因如此,全球监管机构都要求生物药企业证明——细胞分裂到计划使用的最大次数时,依然保持安全、稳定、合格。这个"最大允许分裂次数",就是体外细胞代龄限值(LIVCA),在部分监管文件里也被称为CAL(Cells at the Limit of In Vitro Cell Age)。
2025年,来自辉瑞、FUJIFILM Diosynth、再生元、诺华、默克的作者代表BioPhorum(BPOG)多公司联盟,在 PDA Journal of Pharmaceutical Science and Technology 发布了一份专门针对LIVCA的行业白皮书。ICH Q5B早就提出了这项要求,但只给了原则性的方向,没有说清楚具体怎么落地,导致各家企业在实操细节上的理解和做法长期存在差异。这份白皮书的价值,就是基于行业问卷调研,把“你要证明这一点”,变成了“业内已经验证过、并获得监管批准的几种可行做法”。
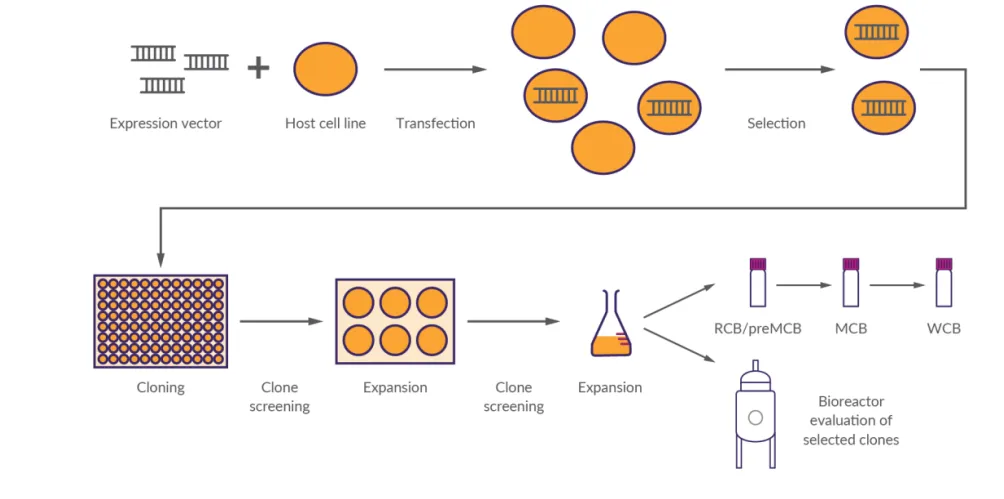
图1 细胞株开发流程图(来源LIVCA的行业白皮书2025)
证明方式:故意把细胞“用到极限”
LIVCA的验证逻辑,是故意让细胞在种子链扩增过程中比正常生产多分裂几代(留出缓冲空间),在这个极限代次上做一整套检测,全部通过才能支撑新药上市申请(BLA)。
这里有个容易被忽略的概念区分:常规生产批次追求的是尽量减少不必要的传代、提高产线利用率;LIVCA验证批次则是故意在摇瓶、波浪袋等环节人为增加传代,模拟最坏工况。两者的产品用途也完全不同——LIVCA验证批次的产品不能用于商业销售或临床,只能用于检测分析。
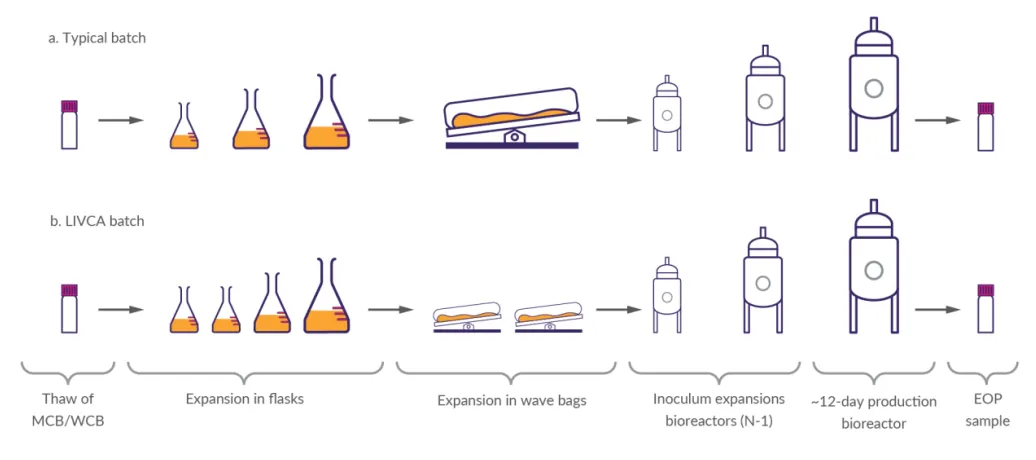
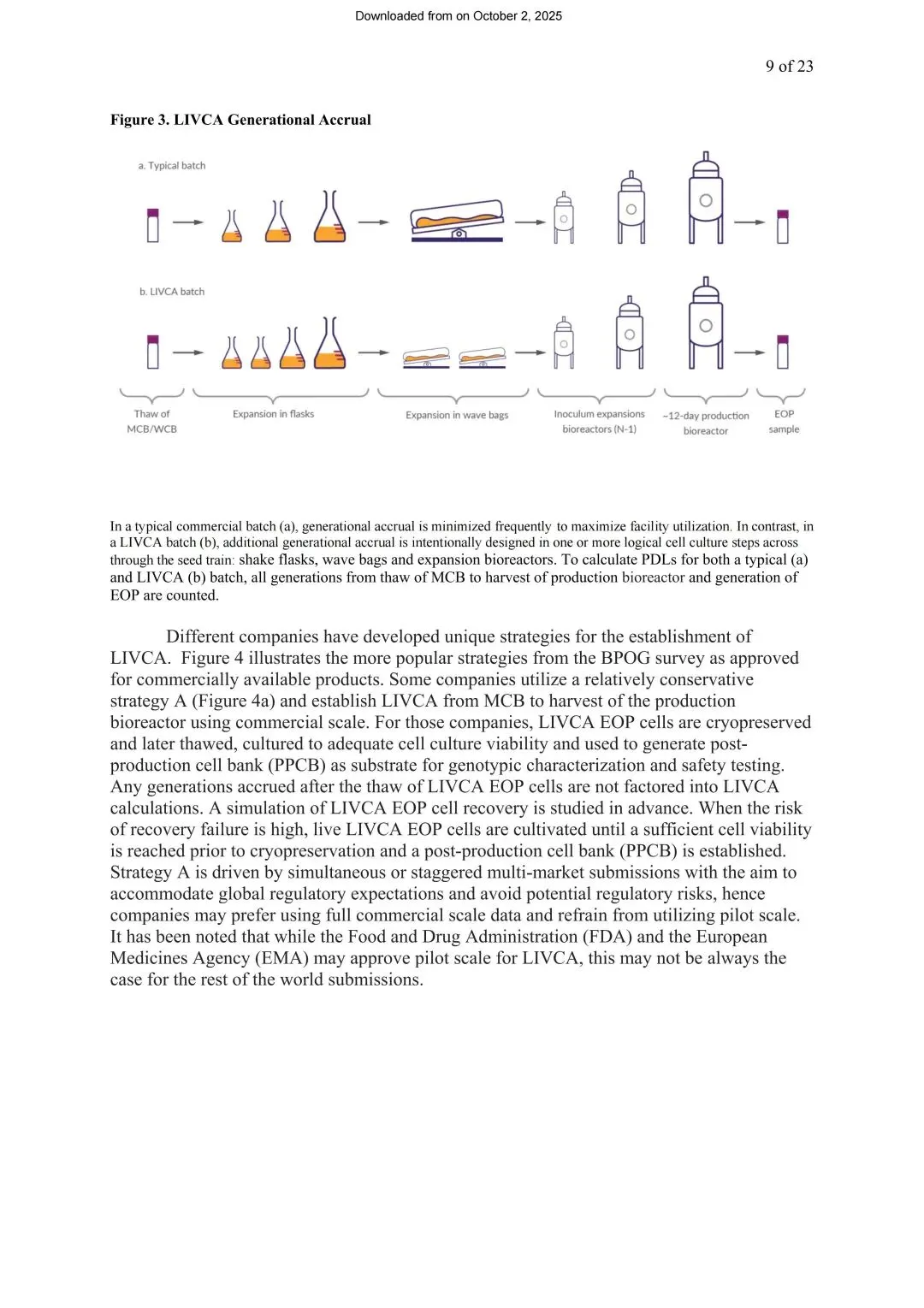
图2 LIVCA代次累积对比图(来源LIVCA的行业白皮书2025)
直观对比常规批次(a)与LIVCA批次(b)从MCB/WCB解冻到EOP样本的完整路径差异
白皮书给出了具体的PDL计算公式。种子链扩增阶段(摇瓶、波浪袋、扩增罐)按公式 x = ln(Nt/NI)/ln(2) 计算,NI是接种密度,Nt是收获密度。生产罐阶段如果工作体积不变,则用Day 0接种密度和峰值密度(而非收获时密度)计算——这一点很关键,因为收获时细胞已进入衰亡期,密度下降不代表分裂减少,用这个数字算会严重低估实际代次。
最容易被漏算的环节是MCB→WCB这一步:白皮书明确写到,WCB通常需要从MCB解冻后再积累10~15代,这部分必须100%计入LIVCA总代次。补料分批工艺里,生产罐阶段通常运行10~14天(有时延长到21天),累积3~6代或更多,相比种子链扩增阶段,这只是总代次中很小的一部分。
白皮书的标准数据包:四大模块
申报LIVCA数据分四块,前两块是法规强制要求:
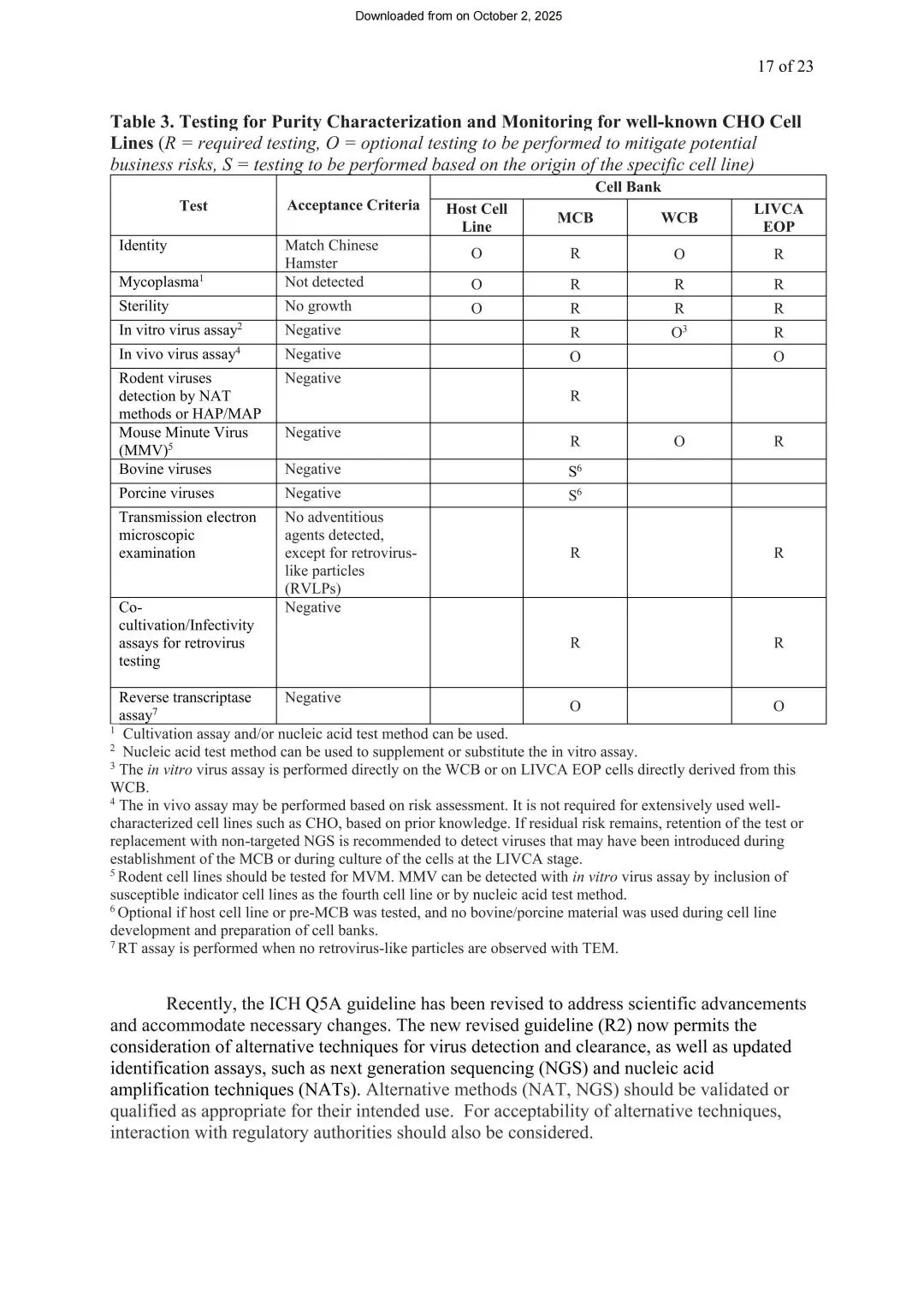
纯度与安全性表征(强制,对应ICH Q5A(R2)、Q5D)
证明MCB、WCB和LIVCA EOP细胞中没有外源污染,潜伏病毒没被激活。表3列出了具体的检测矩阵,包括细胞鉴别、支原体、无菌检查、体外/体内病毒检测、啮齿动物病毒(含MMV)、透射电镜、逆转录病毒共培养检测、逆转录酶实验等,并按MCB/WCB/EOP分别标注了“必做”和“可选”。
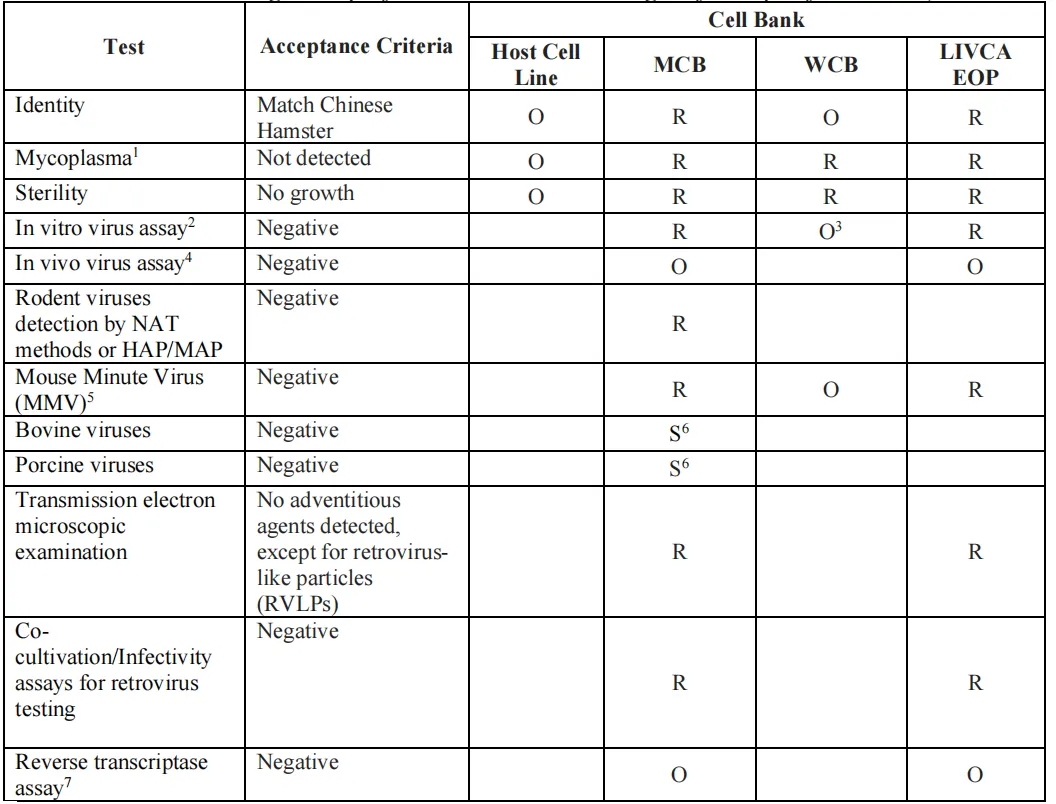
图3 已知CHO细胞系纯度表征与监测检测(来源LIVCA的行业白皮书2025)
白皮书特别提到,监管期望企业采用正交方法评估安全性——也就是用多种独立检测手段互相印证结论,而不是依赖单一方法,这样即使某一种方法存在局限,其他方法也能补上。值得注意的是,ICH Q5A(R2)修订后,已经允许用NGS、NAT等新方法作为传统检测的替代或补充方案,比如用非靶向NGS检测可能在MCB建立或LIVCA培养阶段引入的未知病毒。
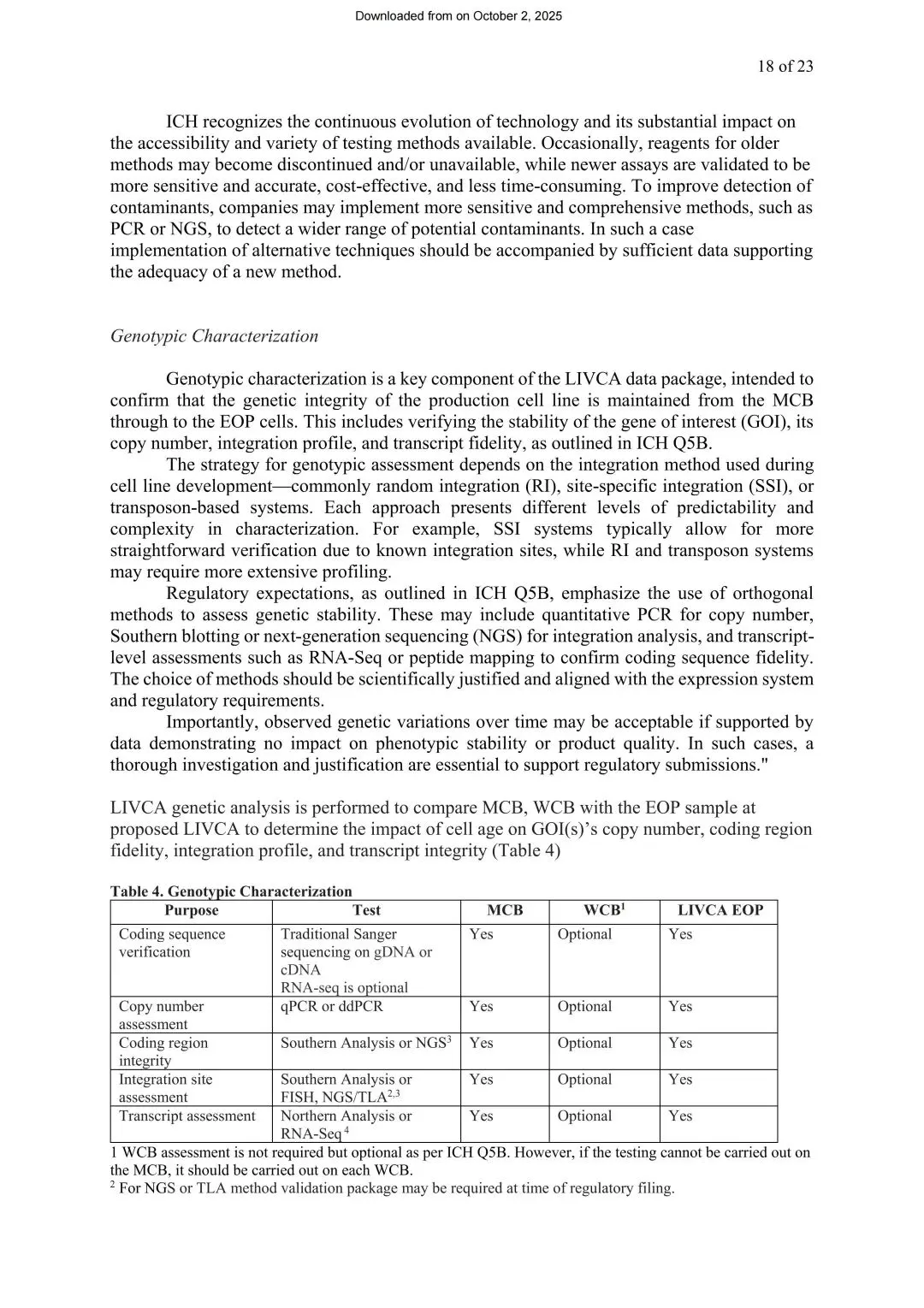
基因型表征(强制,对应ICH Q5B)
证明目的基因(GOI)的拷贝数、整合谱、转录保真度在传代中保持稳定。白皮书给出以下方法矩阵:
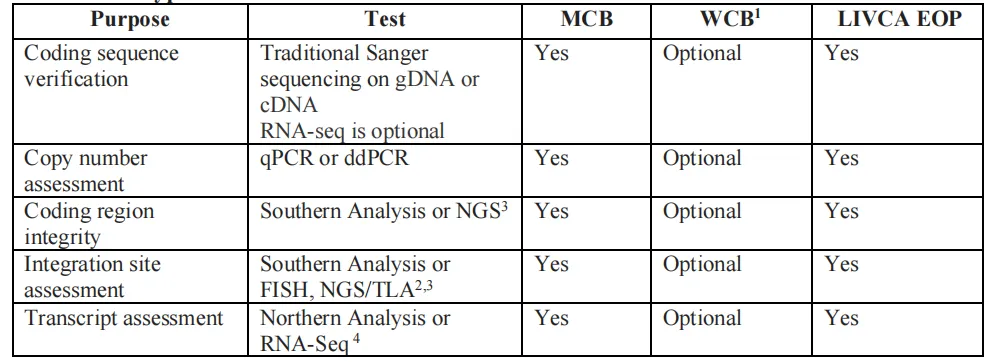
图4 基因型特征分析方法(来源LIVCA的行业白皮书2025)
同样强调正交验证的思路——NGS不是要取代Southern blot等传统方法,而是作为多种方法互相验证的一环,具体怎么选要基于科学依据,并与表达系统、监管要求相匹配。另外原文也写明:WCB检测并非强制要求,只有当MCB无法完成检测时才需要对每批WCB做检测。
还有一个之前容易被忽略的点:白皮书指出基因型评估策略其实取决于细胞株开发阶段使用的整合方式——随机整合(RI)、定点整合(SSI)或转座子系统。SSI因为整合位点已知,验证相对直接;而RI和转座子系统由于整合位点不可预知,往往需要更广泛的图谱分析。行业里最常见的CHO细胞株恰恰多是随机整合,这类细胞系在整合位点分析上对检测灵敏度和覆盖度的要求会更高。
表型表征(行业通用,非法规强制)
监测VCD、细胞活率、生长速率/倍增时间、培养滴度、比生产率(qP)这些“工作状态”指标,确认LIVCA批次的细胞培养表现和历史商业批次一致。
产品质量表征(核心审评要点)
证明做出来的蛋白本身没变质,包括鉴别(肽图等)、效价(细胞生物学活性)、纯度与杂质(RP-HPLC、SEC、HILIC糖基化谱、电荷变异体分析、宿主细胞蛋白残留等)一系列分析。
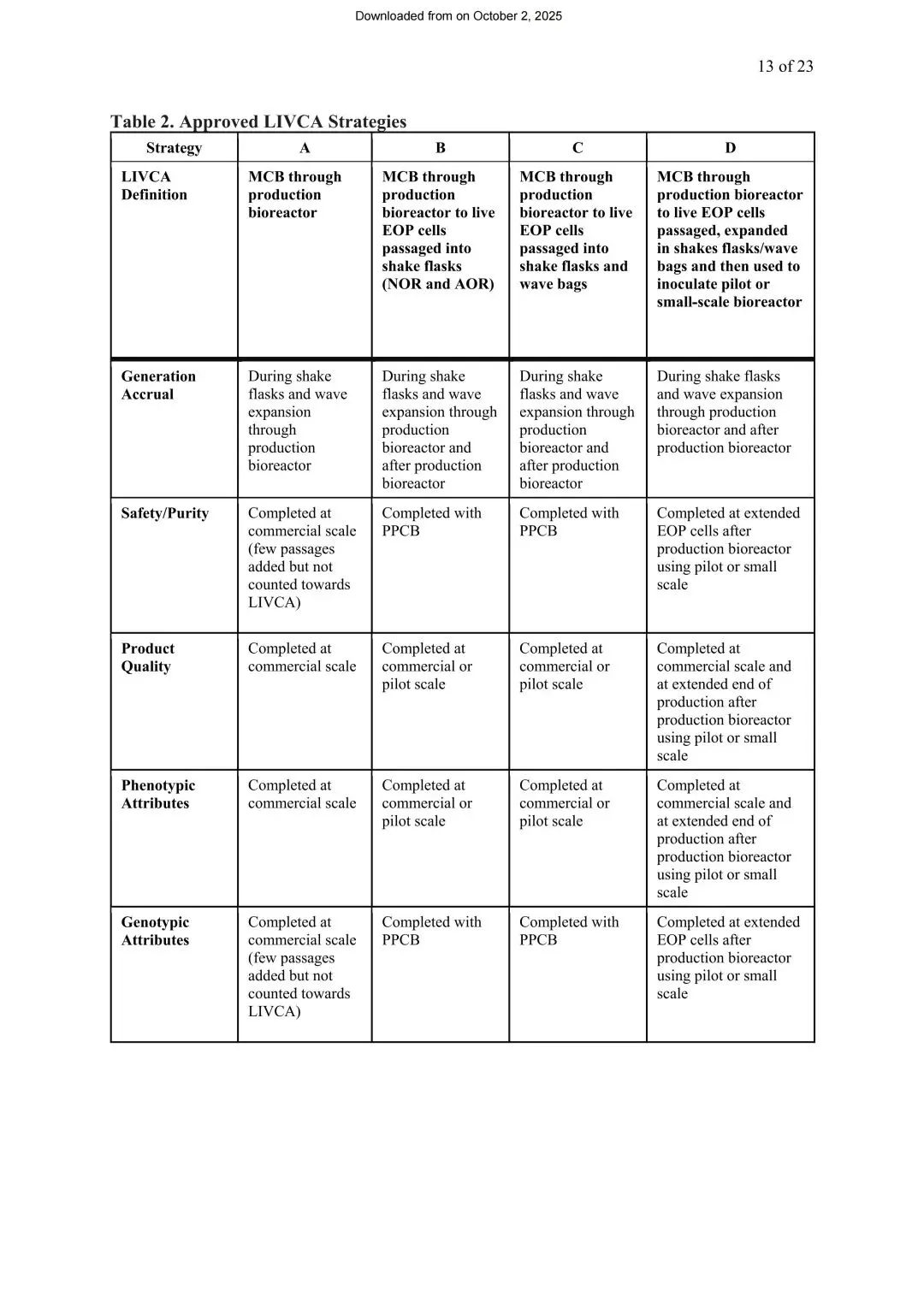
四种实施策略,企业可以按自身节奏选
白皮书基于BPOG行业问卷,总结了全球已获批的四种LIVCA实施路径,全部已通过监管审批并获得商业化许可:
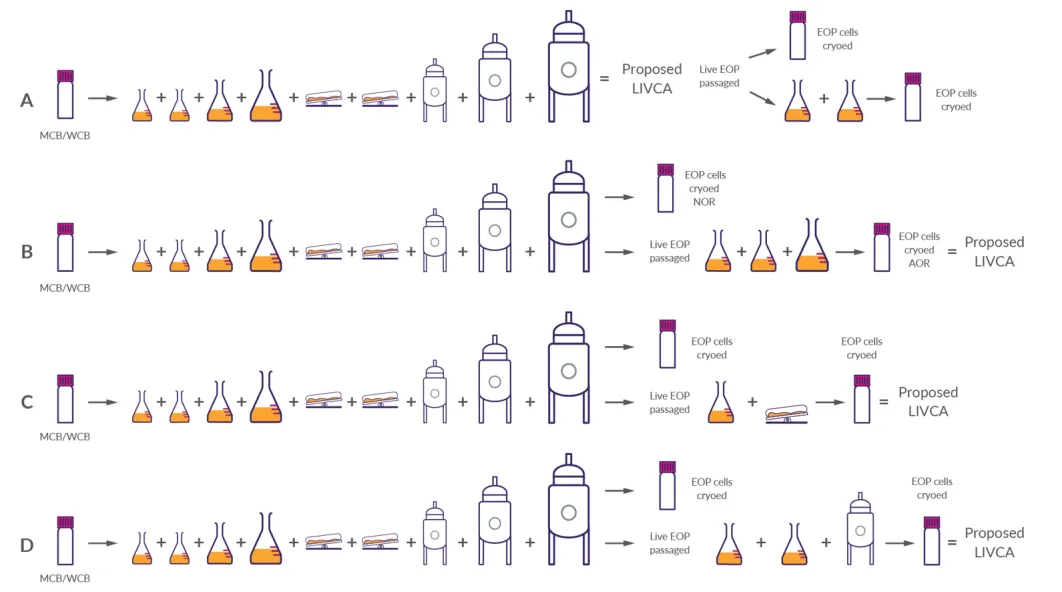
图5 实施策略(来源LIVCA的行业白皮书2025)
策略A(保守型):LIVCA严格定义为MCB到生产罐收获,不含收获后的任何传代。安全性和基因型检测都在商业化规模完成。这种策略监管认可度最高,但完全没有缓冲空间,多数受同步多市场申报驱动选用。
策略B(双区间):在策略A基础上划分“常规运行范围(NOR)”和“可接受运行范围(AOR)”,AOR覆盖EOP细胞收获后在摇瓶中继续传代5~10代以达到可用的细胞活率,制成PPCB(产后细胞库)用于基因型和安全性检测。
策略C(产能友好型):商业化批次正常运行、产品质量数据来自商业批次,活的EOP细胞收获后在摇瓶/波浪袋中继续传代积累更多PDL,制成PPCB做基因型和安全性检测。
策略D(行业首选):在策略C基础上,把扩增后的EOP细胞继续接种到中试或小试规模生产罐,完整复刻商业工艺生成新的EOP样本,用于LIVCA表征。这样既能完全释放商业化产能,又能让产品质量和表型数据分别来自商业批次和中试批次两个维度。
不管选哪种策略,最终都要靠基因型和安全性数据兜底。策略决定在哪个阶段、用什么规模做实验,但实验本身的精度和深度,决定了申报材料能不能扛住监管问询。
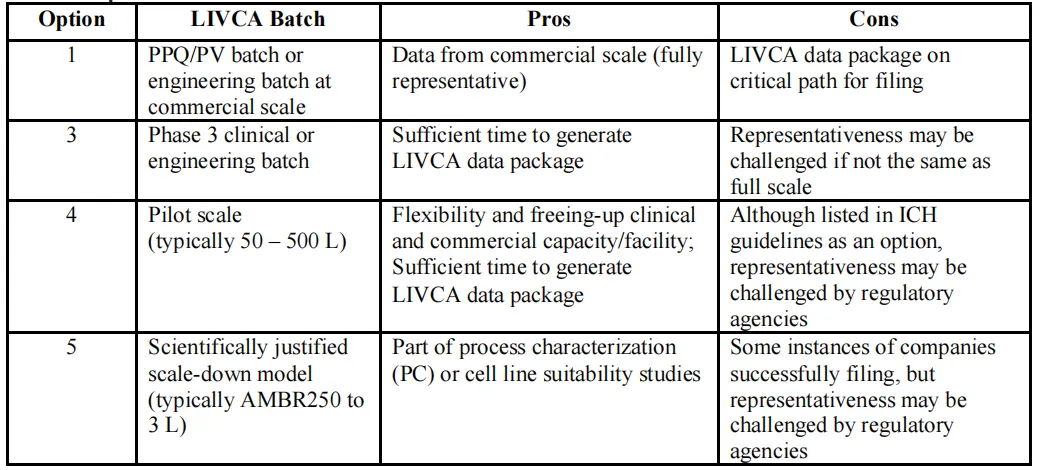
白皮书还总结了五种可选的LIVCA实施规模,从代表性最强但占用关键申报路径的"PPQ/工程批次”,到灵活性最高但监管认可度最低的“科学验证缩小模型(AMBR250~3L)”,企业可以根据项目阶段、申报市场和产能压力灵活组合。
舒桐能提供的技术路线:按LIVCA全流程匹配
把白皮书的检测要求按LIVCA实际执行的时间线展开——从MCB到WCB、种子链扩增、生产罐、最终到EOP——可以看到不同阶段需要的检测内容差异,而基因型表征相关的核心检测几乎都集中在EOP阶段完成。
四条路线分别从整合位点、全基因组、转录本、病原体安全性四个维度切入,覆盖白皮书基因型表征和安全性表征模块要求的核心检测项,也契合白皮书反复强调的“正交验证”思路——多种独立方法互相佐证,而不是单一方法简单验证。
LIVCA研究横跨种子链扩增、极限代次模拟、基因型与安全性检测,时间和数据质量要求都很高。舒桐在CHO细胞整合位点分析和遗传稳定性检测上已经有过多个IND/BLA阶段的项目积累,协助客户成功获得申报经验。
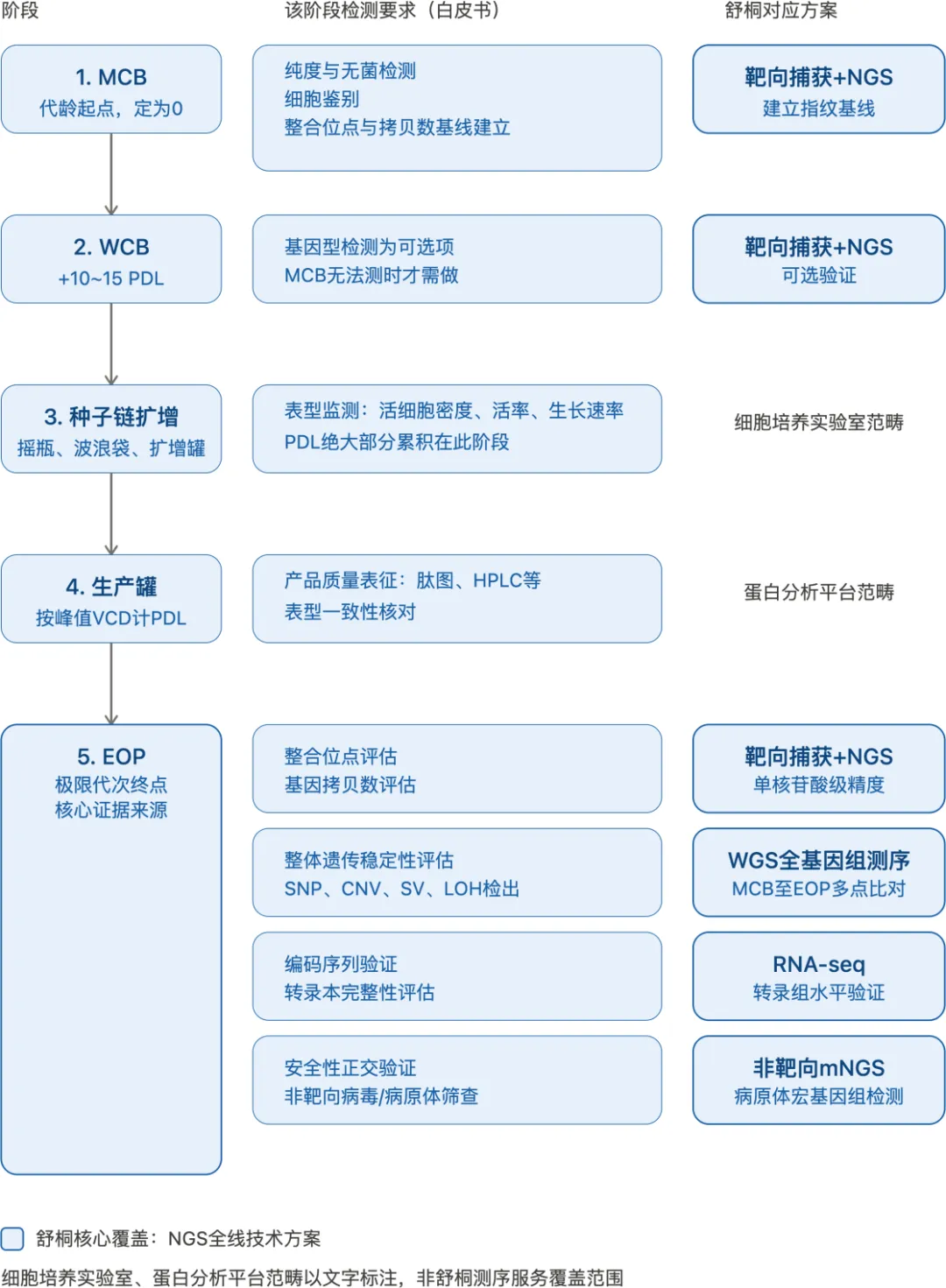
图6 舒桐NGS检测解决方案
上下滑动查看白皮书原文



















关于舒桐