
医药研究

本篇报告是国泰海通证券政策和产业研究院核酸药物系列深度报告的第二篇——心血管专题。siRNA在心血管慢病赛道发展态势势如破竹,国产企业全面布局,部分领域全球领先,产业前景广阔。
摘要
Summary

目录
Summary


本篇报告是国泰海通证券政策和产业研究院核酸药物系列深度报告的第二篇——心血管专题。小核酸药物在心血管领域进展速度较快,在罕见病FCS、ATTR等适应症上进行充分验证后,迅速拓展至慢病领域,目前Inclisiran(PCSK9 siRNA)已上市,另有多个管线正积极开展临床试验,包括靶向Lp(a)的核酸药物已有3款处于临床III期;靶向APOC3的核酸药物已有2款正在开展针对严重高甘油三酯症患者的III期临床;靶向AGT的核酸药物已有1款正在开展高血压III期临床试验;此外,双靶点siRNA亦是下一个热点研发方向,心血管领域已有3款进入临床阶段,进度最快的管线目前处于I/II期。中国企业在小核酸药物研发方面表现领先,不仅在降脂、降压、血栓、双靶点等领域全面布局,在凝血领域甚至全球领先,其余领域亦处于加速追赶状态。心血管是小核酸药物第一个验证的慢病领域,值得进一步跟踪研发进展和上市后销售额爬坡情况。
1. 降脂:市场大有可为,国产加速追赶
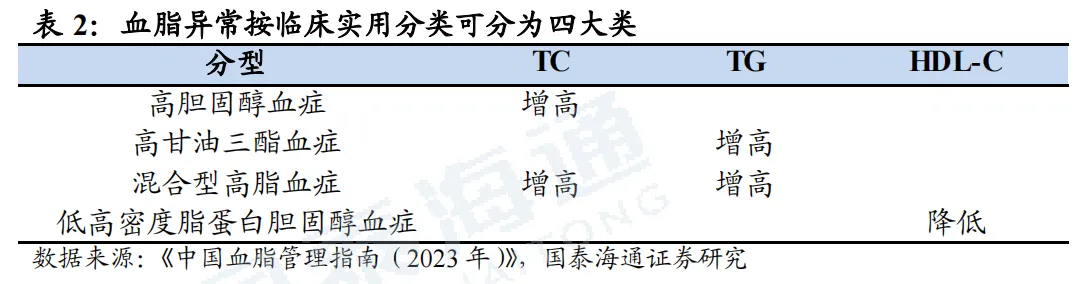
1.1. 血脂异常市场空间广阔,存在较大未被满足需求
1.1.1. 血脂异常是ASCVD的危险因素之一,全球及中国患病人数庞大
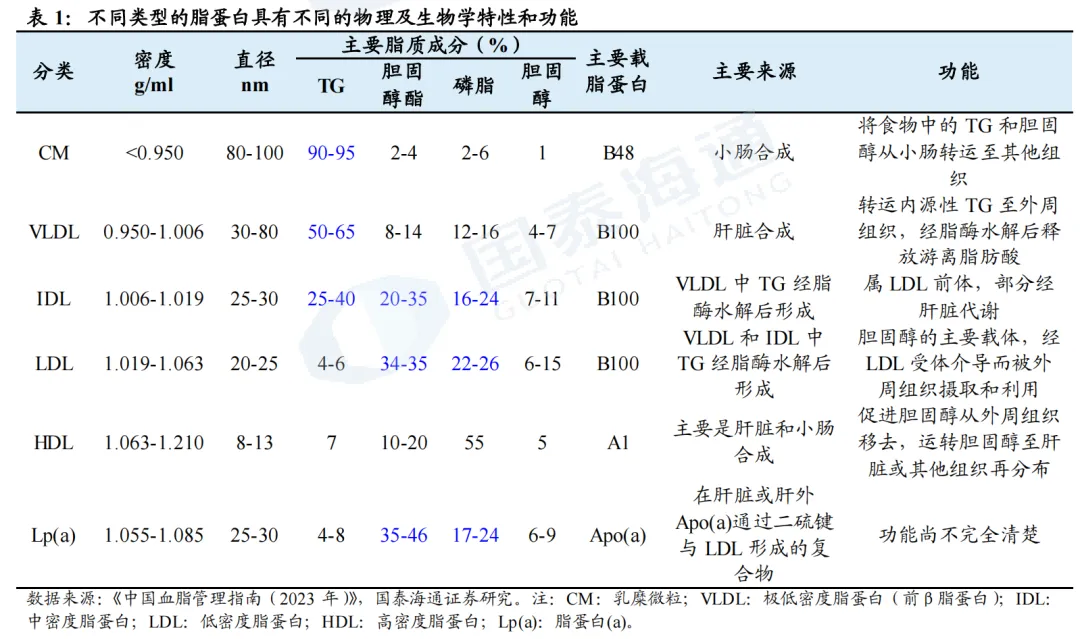
血脂是血清中胆固醇、甘油三酯(TG)等的总称。血脂不溶于水,必须与载脂蛋白(Apo)结合形成脂蛋白才能溶于血液,被运输至组织进行代谢。脂蛋白分为乳糜微粒(CM)、极低密度脂蛋白(VLDL)、中间密度脂蛋白(IDL)、低密度脂蛋白(LDL)、高密度脂蛋白(HDL)、以及Lp(a)。
降低LDL-C
降低TG
混合型血脂异常
合并基础慢病的血脂异常高危人群
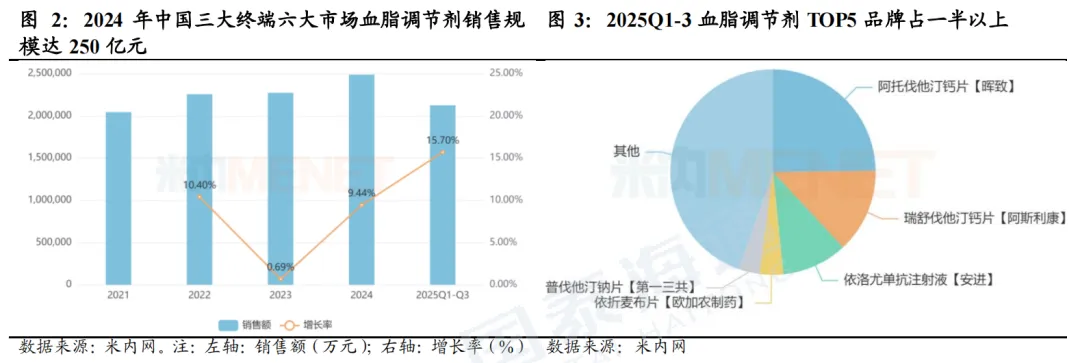
2024年中国血脂调节剂市场达250亿元。根据米内网,2024年中国三大终端六大市场血脂调节剂(化药+生物药)销售额约250亿元人民币,增速达9.44%,2025Q1-3销售额亦超过200亿元,前五大品牌分别为阿托伐他汀钙、瑞舒伐他汀钙、依洛尤单抗、依折麦布、普伐他汀钠。
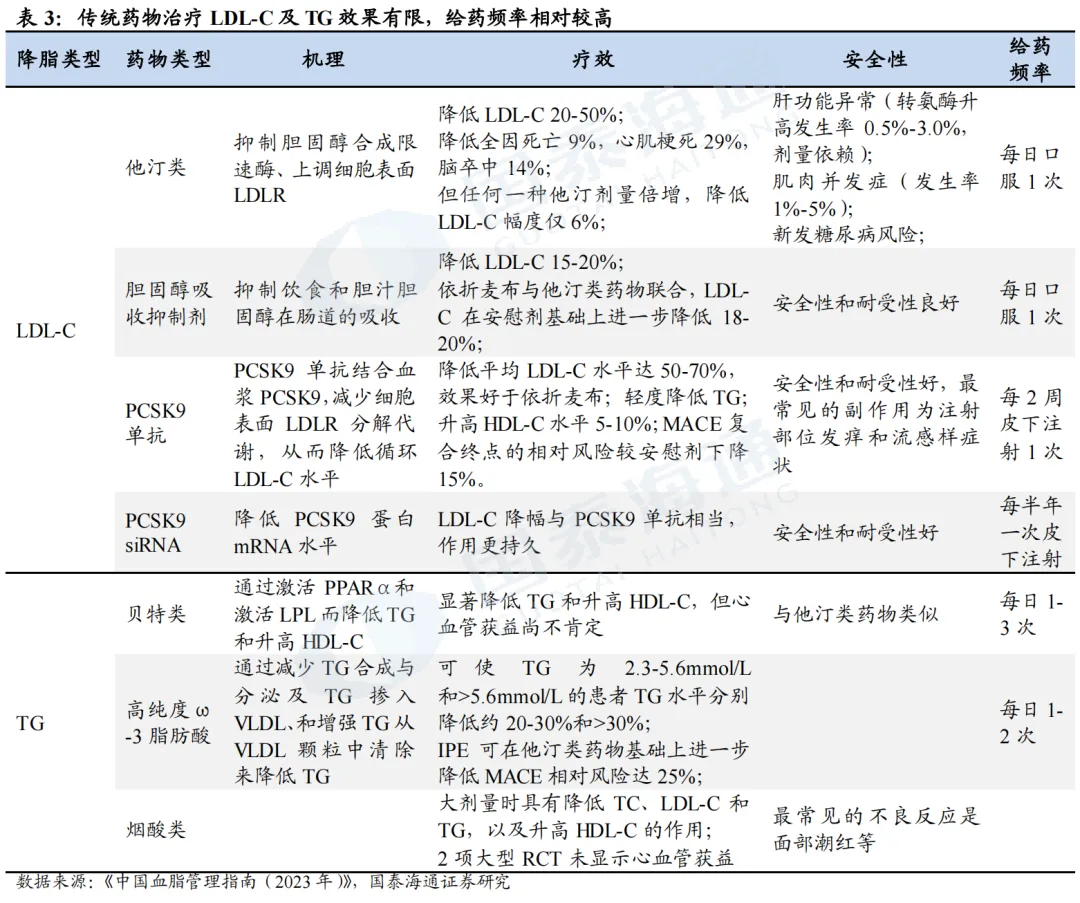
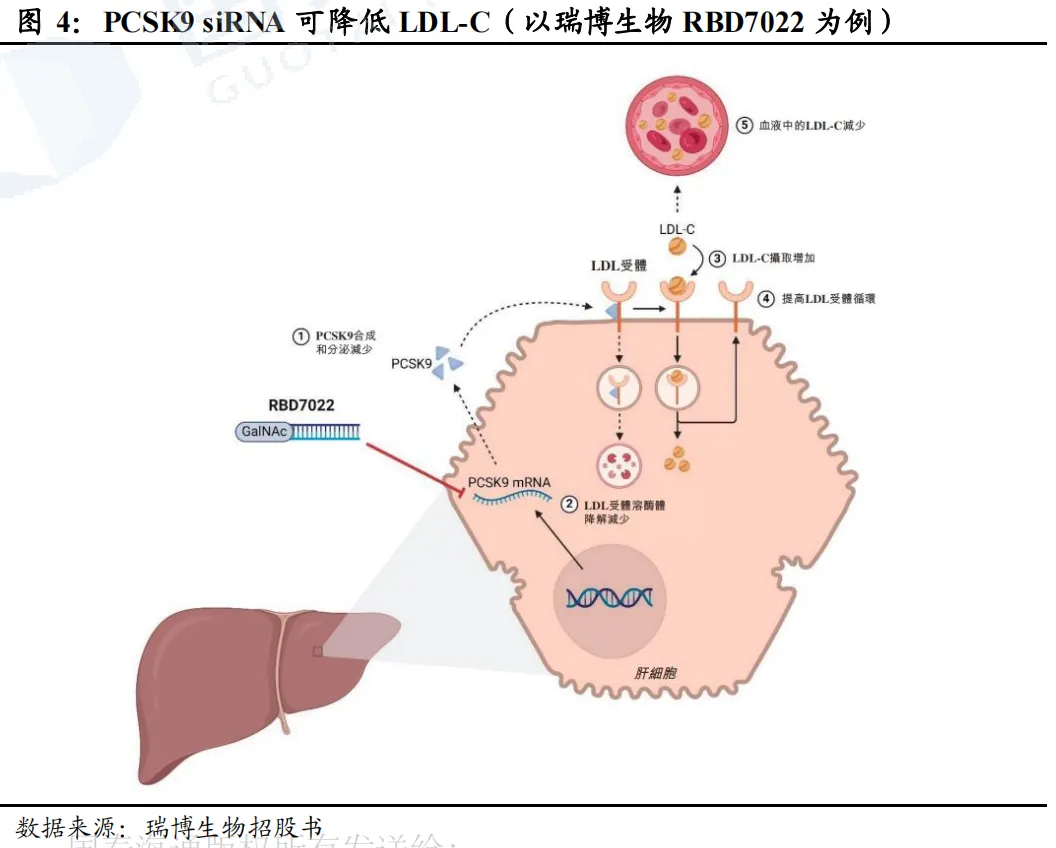
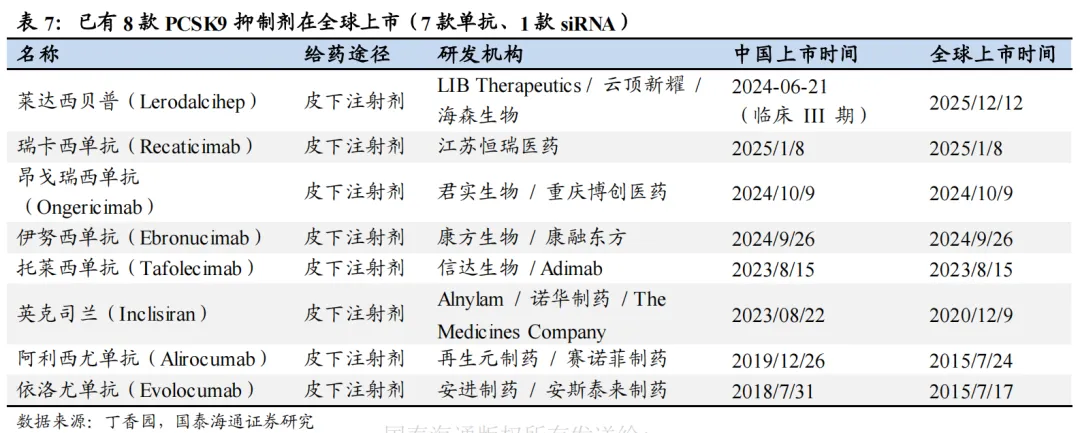
PCSK9靶点已验证,多个PCSK9抑制剂相继开发上市。2003年,科学家发现PCSK9基因的功能获得型突变会导致家族性高胆固醇血症(FH)。PCSK9通过与LDLR结合,促进LDLR在溶酶体中的降解,减少肝细胞表面LDLR的数量,从而降低LDL-C的摄取和清除,增加血液中LDL-C水平。通过抑制PCSK9,则可减少血液中LDL-C的水平,后续多个抑制PCSK9的药物被相继开发。2015年,PCSK9单抗(Alirocumab、Evolocumab)获批;2020年,PCSK9-siRNA药物Inclisiran获批;2023-2025年,多个国产PCSK9单抗亦获批上市。同时,多个口服给药PCSK9抑制剂处于临床试验或即将上市阶段(如默沙东MK0616环状多肽、诺和诺德NN6434等)。
1.2.1. Inclisiran(Alnylam/诺华)成功商业化

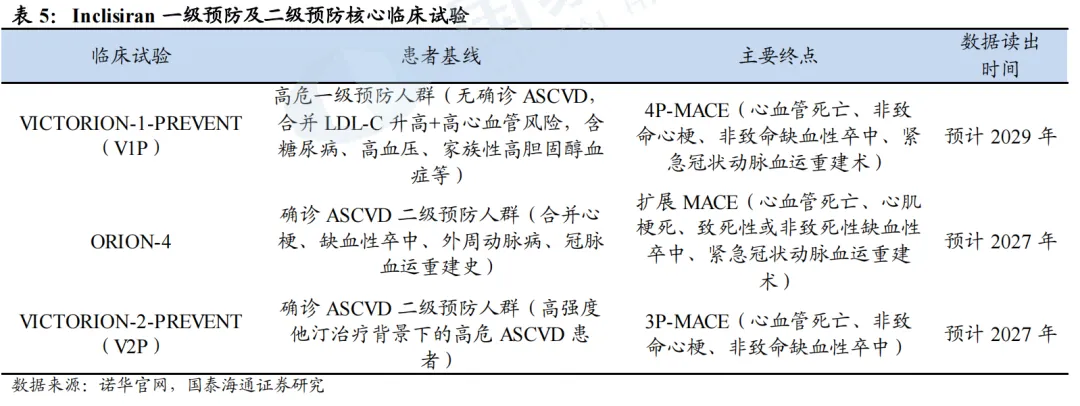
(4)V1P、ORION-4、V2P
此外,Inclisiran还正在开展三项关键临床研究,不再以LDL-C下降等间接指标作为评价标准,而是直接通过心肌梗死、卒中等MACE(主要不良心血管事件)指标,来验证Inclisiran能否为不同风险层级的人群带来真实的临床获益。
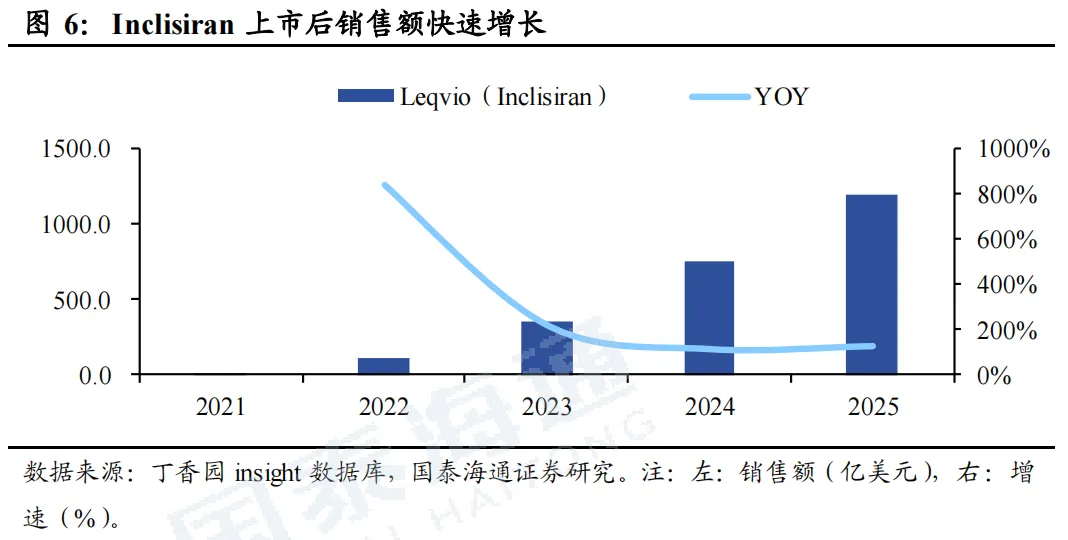
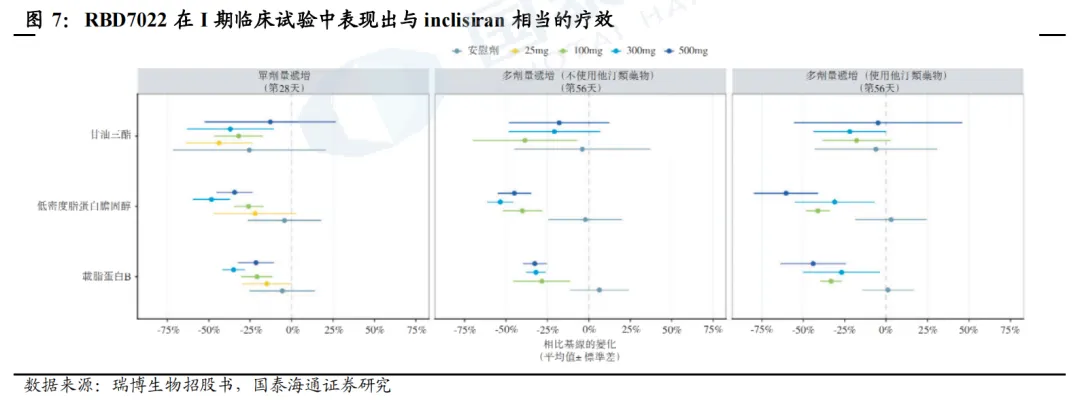
石药和瑞博PCSK9 siRNA进度较快。目前,全球仅Inclisiran一款PCSK9 siRNA上市,且疗效、安全性均获得较大成功。全球进度最快的为石药集团SYH2053和瑞博生物RBD7022,分别于2026年2月、3月登记了III期临床。石药集团尚未披露临床数据。瑞博生物在2025年ESC年会上展示了RBD7022的1期临床试验结果,在使用或未使用他汀类药物的患者中,表现出与Inclisiran相似的LDL-C降低效果,PCSK9水平最大降幅可达75%,且在6个月随访中仍然维持,并且具有良好的安全性和耐受性。2023年12月,瑞博生物以7亿元人民币的首付款和里程碑付款、及最高两位数的特许权使用费率授予齐鲁制药在中国内地、中国香港、中国澳门开发、生产和商业化RBD7022的权利。
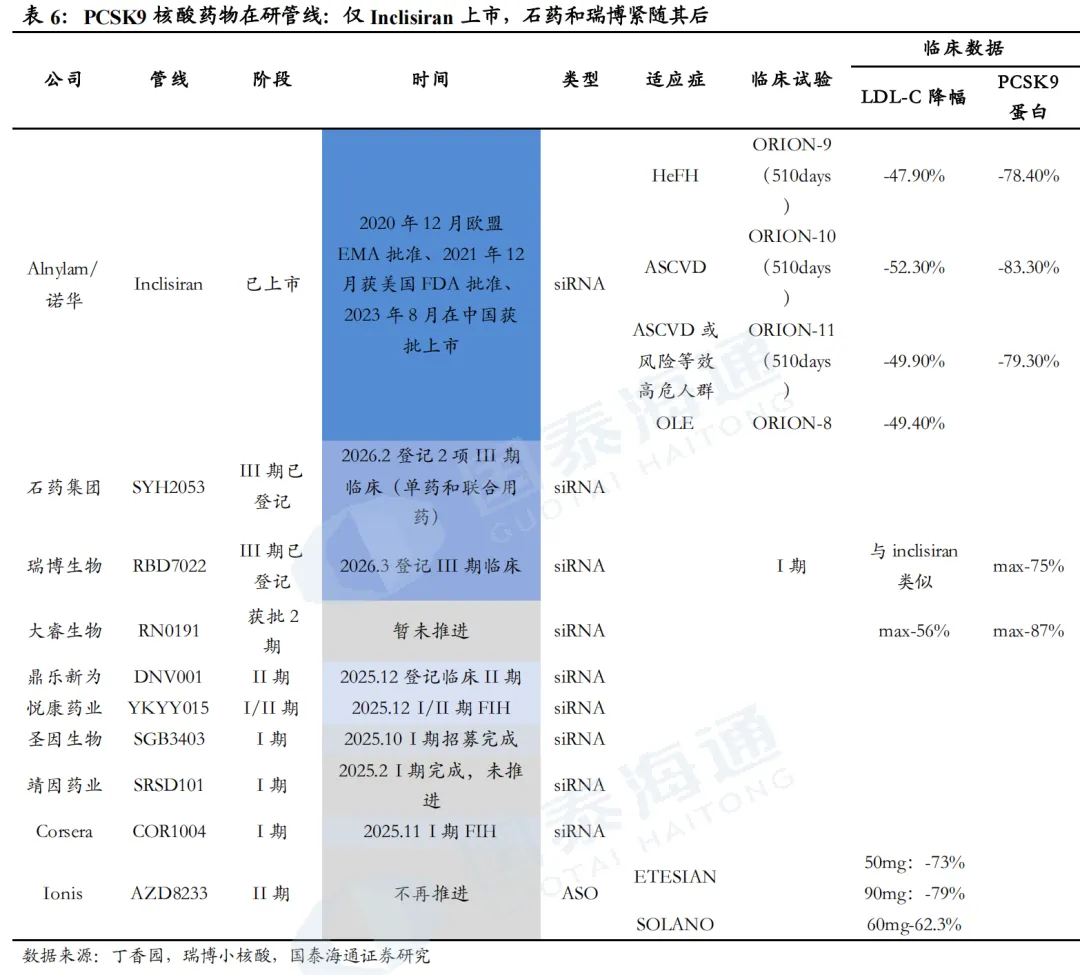
1.3. Lp(a):尚无产品上市,3项大III期临床试验正在进行
1.3.1. Lp(a)成为降脂新靶点,传统疗法作用有限
Lp(a)成为降脂治疗新焦点。研究显示,即使在有效降低LDL-C等主要脂质成分的情况下,Lp(a)仍是ASCVD的独立危险因素,降低50mg/dL的Lp(a)可减少20%的CVD风险,因此,Lp(a)成为除LDL-C等传统降脂治疗靶点外的新焦点。
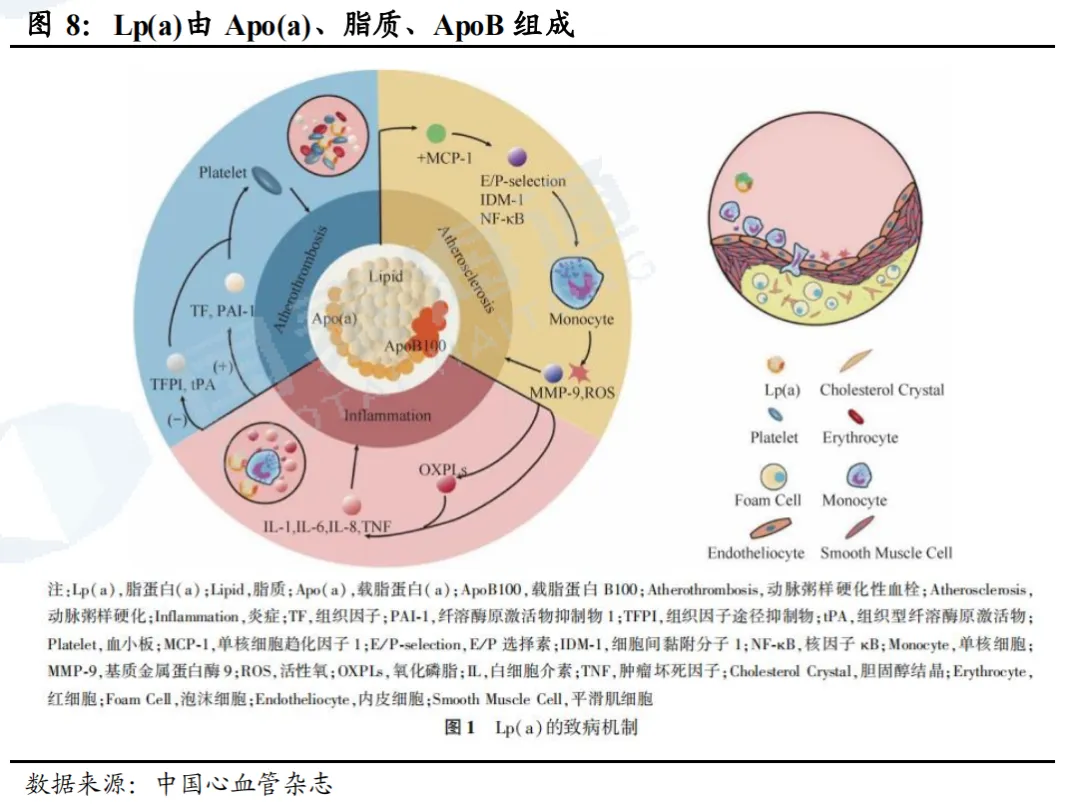
Lp(a)主要致病机制包括三大方面。Lp(a)主要由Apo(a)(载脂蛋白a)和ApoB100(载脂蛋白B100)以及脂质成分构成,Lp(a)水平与Apo(a)亚型大小呈负相关,而Apo(a)亚型大小主要由KIV2结构域重复次数和LPA基因区域单核苷酸多态性共同决定,说明Lp(a)存在个体及种族差异。尽管Lp(a)水平受基因调控的程度不尽相同,其主要的致病机制包括3个方面:(1)Lp(a)具有促血栓形成作用,Apo(a)与纤溶蛋白酶原具备相似的KIV结构域,可以竞争血小板上纤溶酶原受体,同时抑制组织型纤溶酶原激活物,促进血栓状态及内膜纤维蛋白沉积。(2)Lp(a)具有致动脉粥样硬化性,Lp(a)包含的脂质成分比LDL-C所含脂质更易氧化,更易导致动脉粥样斑块形成。(3)参与炎症反应:高浓度Lp(a)可以降低氧化磷脂清除酶的数量及催化效率,促进氧化磷脂形成,从而促进炎症反应。以摩尔计,Lp(a)的致动脉粥样硬化效力约为LDL的5-6倍,这主要得益于其富含OxPLs以及显著的促炎潜力。
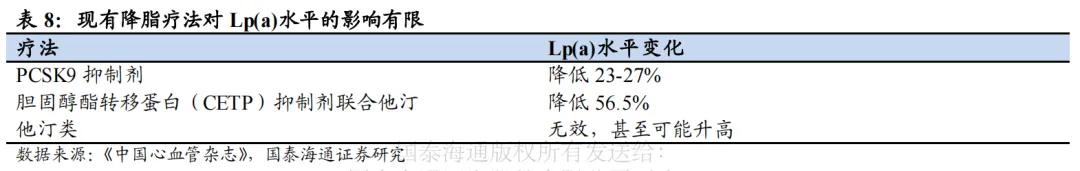
Dicerna/礼来的Lepodisiran(siRNA):通过抑制Lp(a)的关键组成部分Apo(a)的合成来降低Lp(a)的水平。2025年3月,礼来公布Lepodisiran的2期临床试验(ALPACA)积极结果,单次给药最高剂量400mg治疗后第60至180天期间内,Lepodisiran使得Lp(a)水平平均降低93.9%,达到主要终点,第30-360天平均降幅88.5%;而在第180天给予第二针400mg后,第30-360天平均降低达到94.8%,且直至第540天仍保持显著降低。安全性相对较好,最高剂量组丙氨酸氨基转移酶(ALT)和天冬氨酸氨基转移酶(AST)超过正常上限3倍的比例分别为6%、1%,相对较低。3期ACCLAIM-Lp(a)已于2024年3月启动。 Arrowhead/安进的Olpasiran(siRNA):同样靶向Apo(a)。OCEAN(a)-DOSE 2期剂量探索试验评估了Olpasiran在已确诊ASVCD且Lp(a)升高的患者当中的有效性和安全性,在36周时,225mg每12周/每24周注射一次Olpasiran,可分别将Lp(a)降低101.1%、100.5%;安全性表现较好,总体发生率与安慰剂组相似。而对完成48周治疗后停药的患者随访,结果表明停药后24周(总第60周)Lp(a)较安慰剂下降幅度仍超过75%,停药后48周(总第84周)降幅仍维持在40-50%。3期临床试验已于2024H1完成入组。 Ionis/诺华的Pelacarsen(ASO):靶向Apo(a)。2期临床试验评估了其在确诊ASCVD且Lp(a)≥60 mg/dL患者的疗效和安全性,各剂量组实现Lp(a)平均降幅为35%~80%,总体安全性良好。目前正在进行3期临床HORIZON,预计2026H1数据读出,2026H2递交新药上市申请。
另有多项处于2期临床。
靖因药业的SRSD216(siRNA):I期数据疗效优异,2026年3月,靖因药业在ACC披露I期数据,各剂量组较基线最大变化为-87.2%~-94.7%,在第7个月时,300mg和600mg剂量组Lp(a)水平的抑制率仍保持在90%以上,说明SRSD216对Lp(a)的降低效果持续稳定。同时,SRSD216也表现出了良好的安全性和耐受性。靖因药业在2025年底在中美同步启动IIa期,预计2026H2公布2期试验中期数据。 赫吉亚/中国生物制药的Kylo-11(siRNA):靶向Apo(a),目前处于临床2期阶段。2025年11月赫吉亚在AHA年会以口头报告形式发表Kylo-11在中国健康人中的I期临床试验,结果显示,单次给药后即可实现血清Lp(a)水平降幅显著且持久,在24周时,各剂量组降幅维持在83.5%-98.4%。在基线Lp(a)为75-200 nmol/L的受试者中,30 mg组(队列2)在第48周时的Lp(a)降幅仍维持在77.6%,75 mg组(队列3)在第44周时降幅维持在88.8%,225 mg组(队列4)在第40周的降幅维持在96.7%。同时,各剂量组均展现出良好的安全性和耐受性。正在开展的II期研究旨在评价Kylo-11在ASCVD合并Lp(a)升高患者中,采用半年一次给药或一年一次给药的有效性和安全性。 Silence的Zerlasiran(siRNA):目前处于临床2期阶段。II期临床共纳入178例动脉粥样硬化性心血管疾病且Lp(a)水平偏高患者,设置多剂量与给药间隔组别,36周随访结果显示各方案经安慰剂校正后Lp(a)平均降幅均超80%,中位最大降幅可达90%以上;药效具备长效特性,治疗60周后仍能维持可观降脂效果。整体用药安全性良好。
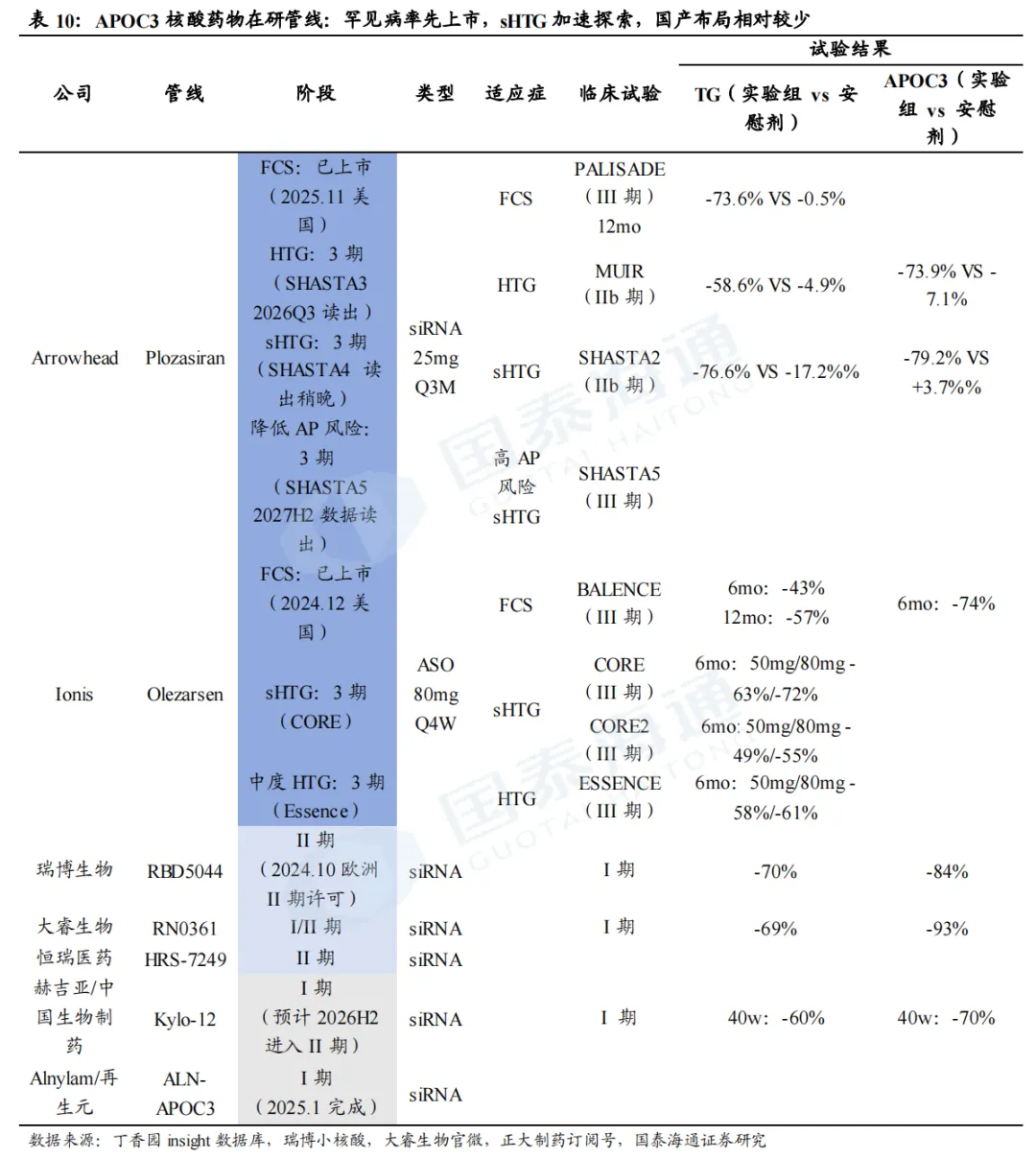
1.4. APOC3:从罕见病到慢病逐步开发,国产布局相对较少
1.4.1. APOC3 siRNA药物探索从罕见病(FCS)到慢病(sHTG)
载脂蛋白C-Ⅲ(APOC3)是调控甘油三酯(TG)代谢的关键蛋白。APOC3一方面抑制脂蛋白脂酶(LPL)活性,阻碍乳糜微粒(CM)、VLDL中的TG水解分解;另一方面阻滞肝脏受体对富含TG的脂蛋白残粒(TRL)摄取清除,双重作用造成血浆TG蓄积升高;反之,通过siRNA沉默肝脏APOC3基因表达,能够解除上述抑制、加速TG水解与残粒清除,实现强效降TG。
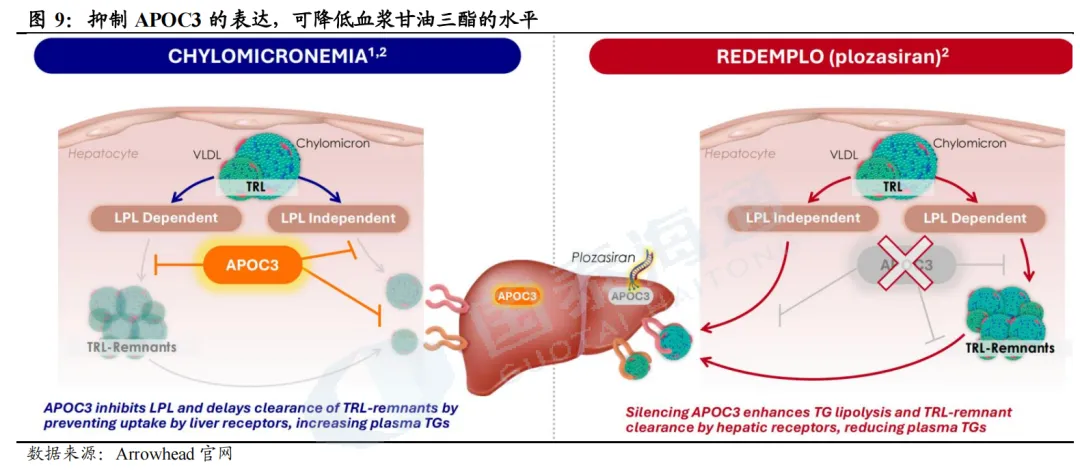
FCS和sHTG患者以高TG为特征。正常人体经肠道吸收膳食脂质后生成乳糜微粒(CM),CM可逐步被肝脏分解、代谢清除;家族性乳糜微粒血症综合征(FCS)因先天基因缺陷,体内CM无法正常降解,后续脂蛋白转化、肝脏清除通路全面受阻,CM大量堆积,诱发极端高甘油三酯,是罕见遗传病;而重度高甘油三酯血症(sHTG)是更广范畴的慢性高血脂疾病。FCS患者以重度高TG、体内乳糜微粒大量堆积为核心特征,极易诱发急性胰腺炎(AP),该并发症致死率达5%~6%;美国临床内分泌医师协会(AACE)、美国内分泌学会(ACE)等多个专业指南推荐将TG降至500mg/dL以下以规避胰腺炎风险。
APOC siRNA药物以罕见病FCS为起点,面向广大sHTG患者人群。根据Arrowhead官网,全美FCS患者约650人(TG≥880mg/dL)、高危sHTG患者约100万(TGs>=880mg/dL或TGs>=500mg/dL且有急性胰腺炎发作史)、全口径sHTG患病人数超300万(TGs>=500mg/dL),重度高TG人群临床未被满足的降脂空间广阔。传统贝特、ω‑3等口服药难以实现强效达标,APOC3靶向siRNA成为针对FCS、sHTG重度高甘油三酯的最优新型治疗方向。
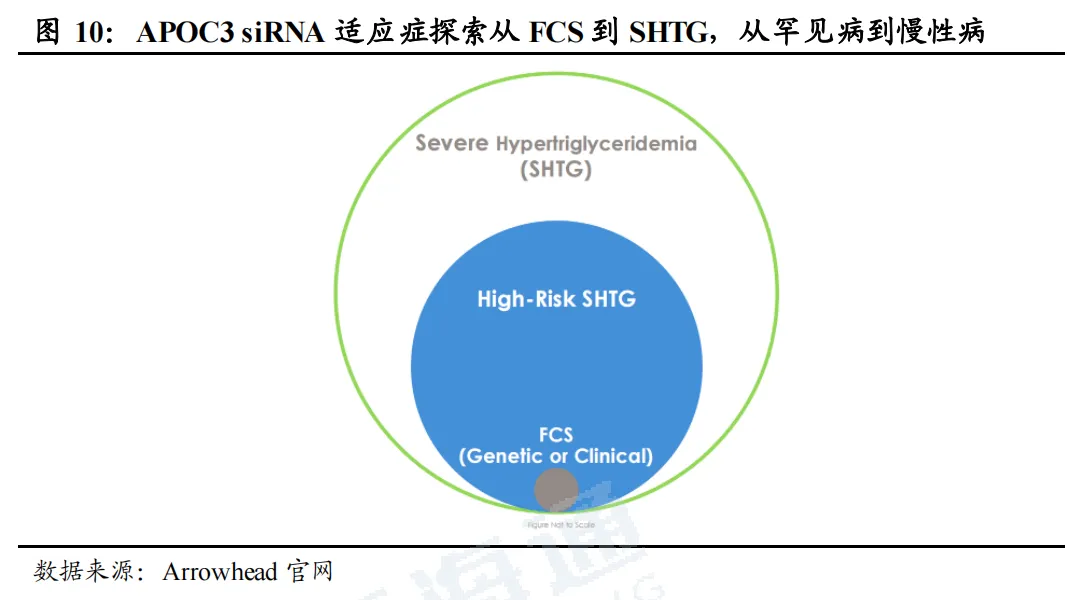
1.4.2. 海外FCS已率先上市,sHTG上市指日可待,国产布局相对较少
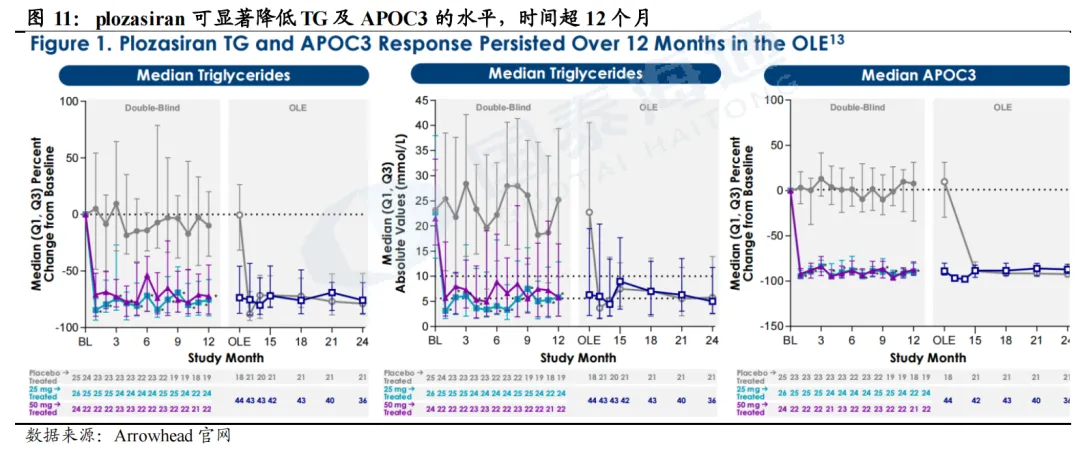
plozasiran(siRNA,Arrowhead)
FCS适应症已于2025年11月在美国批准上市(25mg Q3M SC)。PALISADE是一项3期临床试验,评估plozasiran(Q3M)在FCS患者当中的长期安全性和有效性,主要终点为12个月TG变化幅度。试验结果显示,安慰剂组TG下降幅度为0.5%,而plozasiran组达到73.6%,在TG变化绝对值及APOC3较基线变化百分比方面亦有显著改善,且在后续的OLE试验中疗效持续,安全性和耐受性良好。
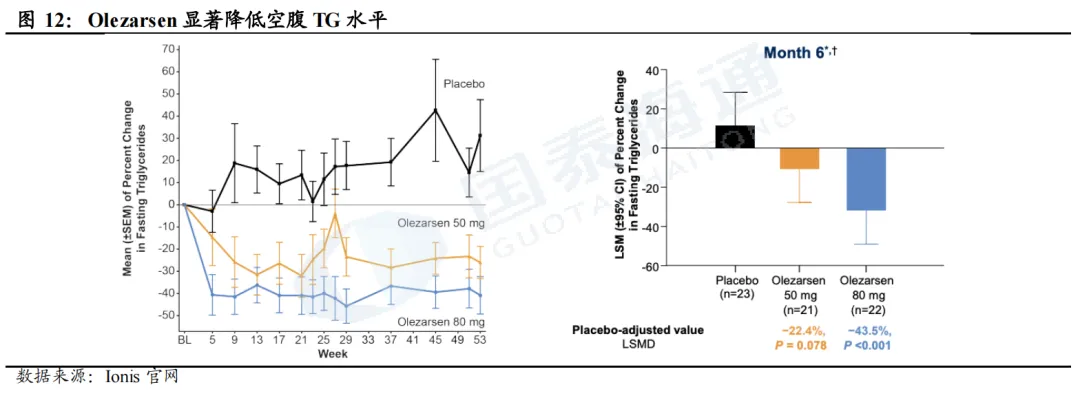
Olezarsen(ASO,Ionis/诺华)
FCS已于2024年12月在美国获批上市(80mg Q4W SC),2025年销售额超1亿美元。BALENCE是一项3期临床试验,用于评估olezarsen在FCS患者当中的疗效及安全性,试验结果显示,在6个月及12个月时,80mg olezarsen较安慰剂分别降低TG水平达43%(p<0.001)和57%;在第6个月,80mg olezarsen较安慰剂降低APOC3水平达74%。同时,olezarsen显示出明确信号可以降低胰腺炎发作(RR=0.10-0.14)。2025年销售额超1亿美元。
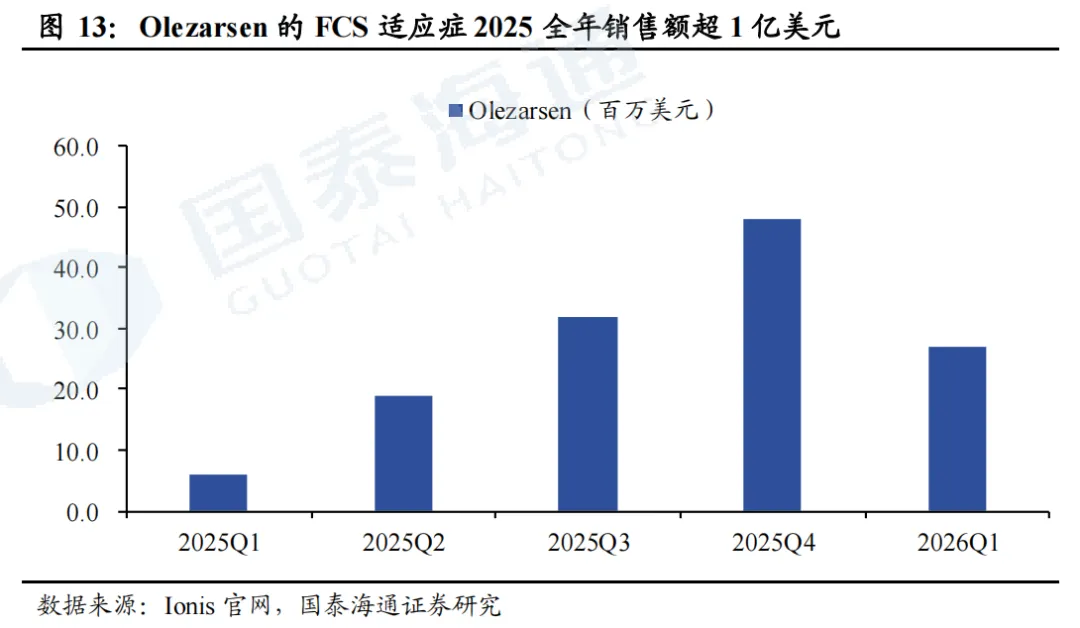
sHTG适应症预计2026年上市。两项3期临床试验CORE和CORE2用于评估olezarsen(50mg/80mg,Q4W,SC)在sHTG适应症的疗效和安全性。在CORE试验中,6个月时,50mg/80mg olezarsen较安慰剂降低TG的水平分别为63%、72%;在CORE2试验中,6个月时,50mg/80mg olezarsen较安慰剂降低TG的水平分别为49%、55%。结果均具备显著性差异(p<0.001)。Ionis目前已向FDA递交sNDA,预计2026年获批上市。根据Ionis投资者交流材料,公司对Olezarsen销售峰值预期超30亿美金。
olezarsen亦被证明可降低中度HTG。Essence研究纳入了空腹TG水平在150-500mg/dL且伴有或存在ASCVD的患者,每月注射50mg/80mg Olezarsen,6个月时,TG水平分别降低58%、61%(p<0.0001),且具有良好的安全性和耐受性。
瑞博生物RBD5044(siRNA):RBD5044是全球第二款进入临床开发阶段的靶向APOC3的siRNA。I期临床结果显示,单次给药即可实现APOC3降低84%,TG下降70%,且在6个月随访中,TG仍稳定维持在基线50%以下,同时,受试者血脂得到全面改善,包括残余胆固醇显著降低达70%,载脂蛋白B(ApoB)降低达20%,以及高密度脂蛋白(HDL)显著升高达40%。RBD5044表现出良好的耐受性,在最高剂量下未观察到剂量依赖性的不良反应活肝酶升高现象,具有成为BIC的潜力。2024年10月,RBD5044获得EMA的II期临床试验许可,目前正加快推进中国II期临床研究。
大睿生物RN0361(siRNA):目前处于I/II期。I期结果显示,RN0361剂量依赖性上呈现出显著的ApoC3与TG抑制作用,持续至给药后第180天,最高达到ApoC3降低93%及TG降低69%。还实现了非高密度脂蛋白胆固醇、极低密度脂蛋白胆固醇及残余胆固醇等显著且持久的降低。
赫吉亚/中国生物制药Kylo-12(siRNA):目前处于I期,预计2026H2进入II期。I期临床结果显示,单次给药10mg即可在40周后仍维持约70%的APOC3和约60%的甘油三酯(TG)降幅,且疗效呈现显著剂量依赖性,预示其同样具备一年给药一次即可实现显著获益的潜力。
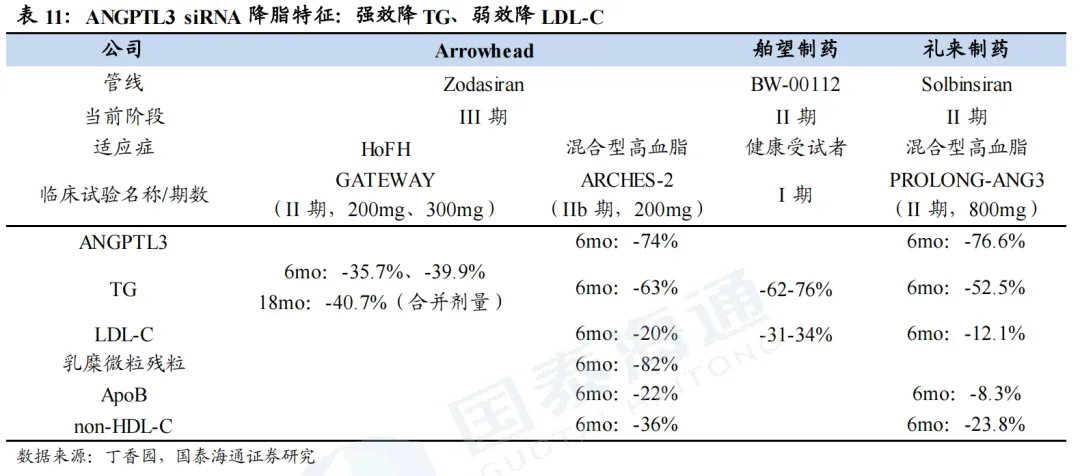
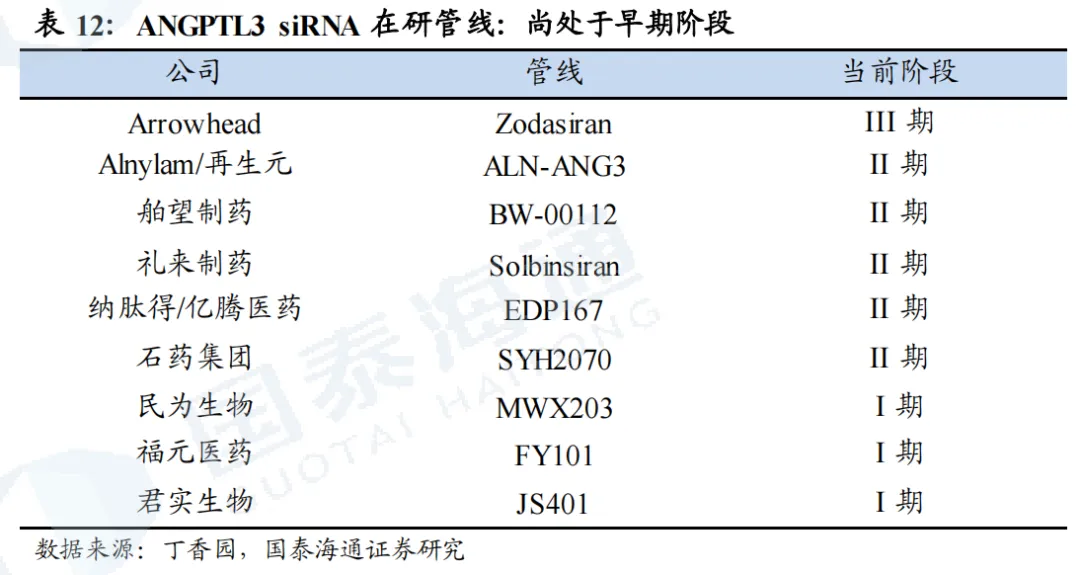
1.5. ANGPTL3:广谱降脂但效果偏弱,竞争格局温和
1.5.1. ANGPTL3 siRNA具备广谱降脂潜力,有望占据利基市场
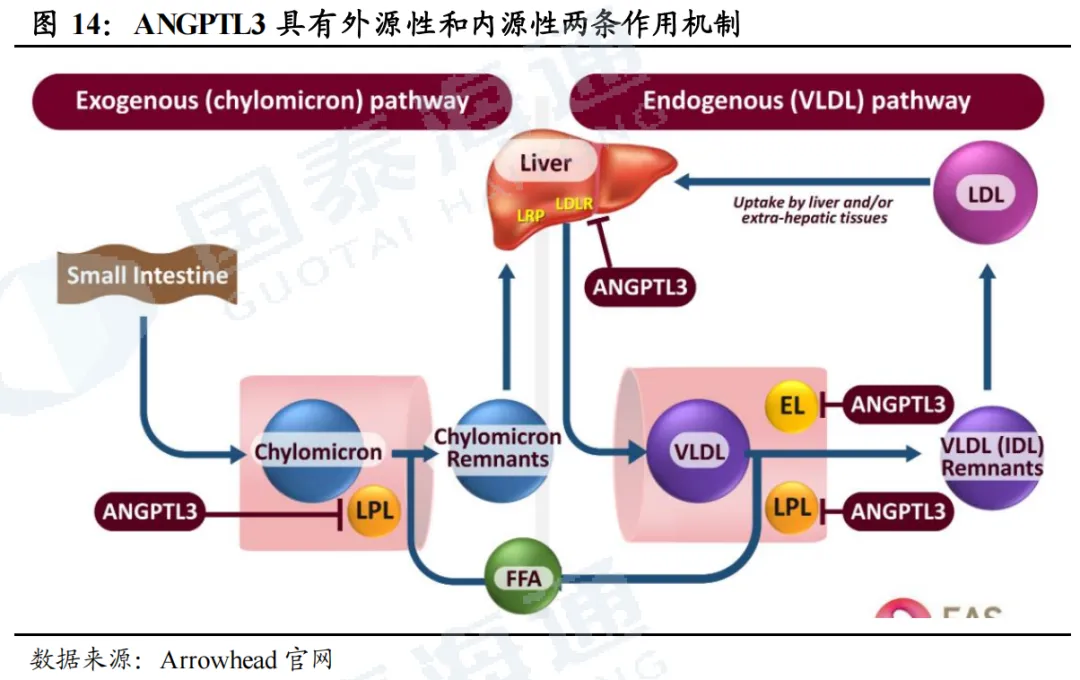
ANGPTL3与多个脂蛋白有关,且靶点安全性高。ANGTPL3(血管生成素样蛋白3)是一种由肝脏分泌的蛋白质,是脂质和脂蛋白代谢的重要调节因子。其作用机制主要为通过抑制两种关键酶来发挥作用,分别是LPL(脂蛋白酯酶)、EL(内皮脂肪酶)。(1)外源性途径(乳糜微粒):小肠吸收食物中的脂肪后,合成乳糜微粒(Chylomicron),用来运输饮食来源的TG;乳糜微粒进入血液后,需要LPL将其内部TG分解为游离脂肪酸(FFA)供身体利用,剩下的颗粒被称为乳糜微粒残粒(Remnants)。如果ANGTPL3活跃,则直接抑制LPL活性,导致TG分解速度变慢,血液中TG和乳糜微粒残粒升高。(2)内源性途径(VLDL):肝脏合成并分泌极低密度脂蛋白(VLDL),用于运输肝脏合成的TG,VLDL同样依赖LPL分解TG,将其转化为VLVL残粒(IDL),后IDL转化为LDL,被肝脏或外周组织摄取代谢。如果ANGTPL3活跃,则同时抑制LPL和EL,阻碍VLDL分解。因此,如果抑制ANGTPL3活性,则可降低TG、LDL-C、VLDL- C的水平。同时,研究发现,先天性缺乏ANGTPL3的人没有已知不良表型,说明抑制该靶点的安全性高。
1.5.2. 尚无ANGPTL3 siRNA推进III期混合型高脂血症临床试验
Zodasiran(siRNA,Arrowhead)
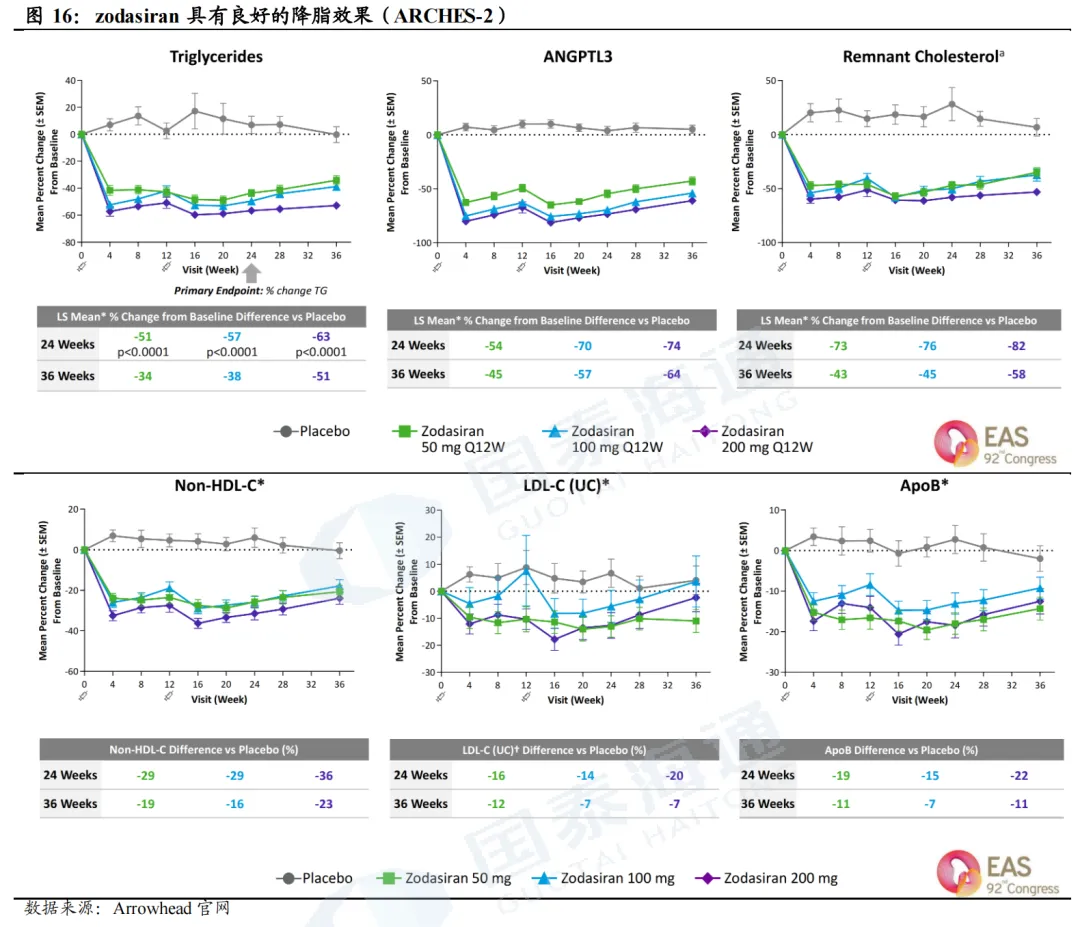
混合型高脂血症IIb期临床试验结果表现良好。IIb期临床试验ARCHES-2结果显示,Zodasiran具有良好的降脂效果和安全性。在第6个月,Zodasiran(200mg,Q12W)可实现ANGTPL3下降74%、TG下降63%、乳糜微粒残粒下降82%、ApoB下降22%、non-HDL-C下降36%、LDL- C下降20%。在OLE试验中,各项指标的降低持续2年以上,包括TG降幅维持在超50%、non-HDL-C降幅维持在30%、APOB降幅维持在超15%。
Zodasiran的HoFH III期临床试验已启动。GATEWAY是一项II期研究,用来评估zodasiran在HoFH患者的有效性和安全性。结果显示,治疗6个月时,200mg、300mg组LDL-C平均下降-35.7%、-39.9%,额外随访12个月,合并剂量组患者LDL-C水平平均下降-40.7%。2025年底,Arrowhead启动了zodasiran 3期临床试验YOSEMITE的首例受试者给药,用于探索在HoFH患者当中的疗效和安全性,主要终点是第12个月空腹LDL- C的变化,公司预计2027年完成试验并递交NDA,2028年上市。
Alnylam/再生元的ALN-ANG3(siRNA):采取差异化布局。1)核心适应症为糖尿病肾病(DKD)伴血脂异常,与Zodasiran差异化竞争。2)II期临床设计采用ALN-ANG3和再生元的ANGPTL3单抗伊维苏单抗进行联用,ALN-ANG3作用于肝细胞内,降解ANGPTL3 mRNA,从源头减少合成;伊维苏单抗则直接结合血液中的ANGPTL3单抗,阻断蛋白功能,从而产生叠加效果。同时,siRNA每三个月一次给药,提供平稳长效的抑制;单抗每月一次给药,提供脉冲式强力清除,联合治疗效果有望增强。II期临床试验于2026年1月启动。
礼来的Solbinsiran(siRNA):目前处于临床2期阶段,混合型高脂血症2期临床试验(PROLONG-ANG3)结果显示,在第180天时,Solbinsiran(800mg,第0/90天皮下注射)可实现ANGTPL3下降76.6%、TG下降52.5%、ApoB下降8.3%、non-HDL-C下降23.8%、LDL- C下降12.1%。
舶望制药的BW-00112(siRNA):已与诺华就BW-00112达成优先谈判权。针对健康受试者的临床1期数据表现良好,TG下降62-76%,LDL-C下降31%-34%。目前,临床II期试验,混合型高脂血症中美同步推进,其中2025年3月中国、2026年1月全球已招募完成;高甘油三酯症已于2024年11月招募完成。
2. 降压:AGT是siRNA潜力靶点,国产布局较多
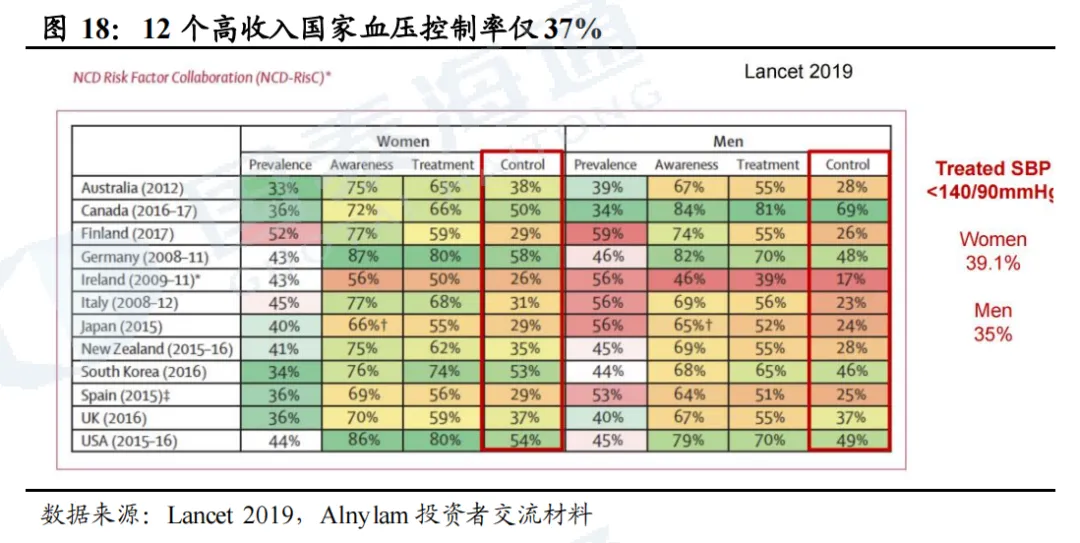
2.1. 全球高血压控制率较低,AGT siRNA潜力较大
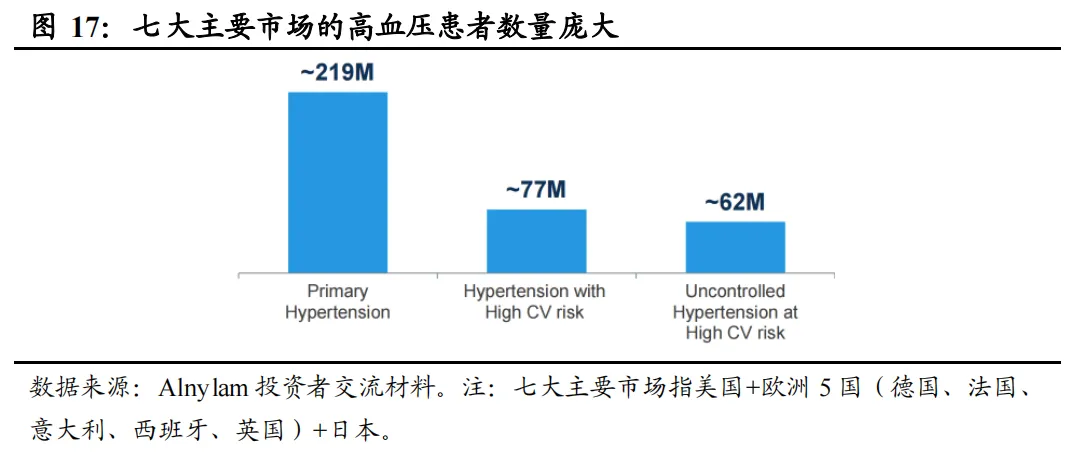
高血压是心肾代谢综合症的主要驱动因素之一,与心功能不全、代谢紊乱及肾功能衰退相互作用。高血压是全球最重要的死亡风险因素,占所有死亡人数的一半以上。根据信立泰招股书,全球及中国高血压患者人数在2024年分别达到15.93亿人、3.53亿人。根据Alnylam,七大主要市场中原发性高血压2.19亿人,有高心血管风险的的高血压患者7700万人,有高心血管风险且未被控制的高血压患者6200万人。
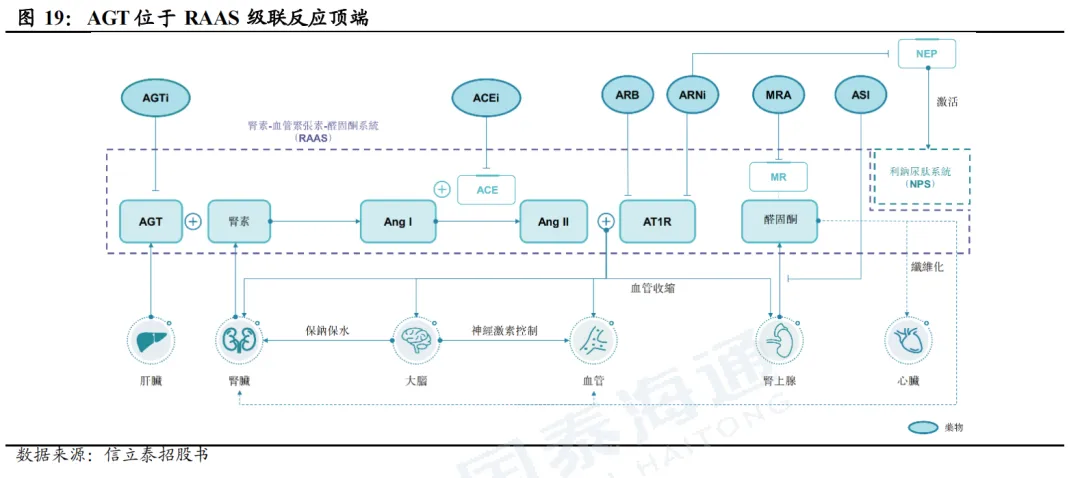
2.2. Zilebesiran进度大幅领先,国产企业布局较多
Zilebesiran(siRNA,Anlylam/罗氏)
Anlylam/罗氏的Zilebesiran是进展最快的靶向AGT的核酸药物(唯一一个进入3期临床)。Zilebesiran在3个2期临床中均表现出较好的疗效。(1)KARDIA1是评估Zilebesiran单药的降压效果,结果显示Zilebesiran可实现长达6个月的血压持续控制;(2)KARDIA2评估Zilebesiran联合1种标准治疗的血压控制效果,与安慰剂相比,在3个月时,Zilebesiran联合不同的抗压药均实现显著血压下降,联合Indapamide、Amlodipine、Olmesartan分别较安慰剂实现平均动态收缩压下降12.1mmHg(p<0.001)、9.7mmHg(p<0.001)、4.5mmHg(p=0.018);(3)KARDIA3评估Zilebesiran联合2-4种降压药治疗用于高心血管风险且血压未控制患者,结果显示,即使患者已使用多种降压药,但在单次注射Zilebesiran 300mg/600mg 3个月后,诊室收缩压(SBP)分别降低5.0mmHg(p=0.04)、3.3mmHg(p=0.18),亚组分析表明,联用利尿剂的亚组患者实现血压降幅更大,300mg/600mg剂量组分别为9.2mmHg、7.0mmHg。同时,Zilebesiran安全性表现良好。
基于KARDIA1/2/3良好的实验设计和数据,Zilebesiran正在推进III期临床。KARDIA1/2/3采取循序渐进的方式,从单药到联合1种药,到联合2-4种药,模拟真实世界从初治到难治的患者情况。基于优异的2期临床数据,Alnylam正在推进Zilebesiran的3期临床Zenith,用于评估其在患有未受控制的高血压且已确诊心血管疾病的成年患者,或尽管接受稳定治疗(至少使用2种降压药,其中1种为利尿剂)后,SBP仍高于140mmHg的患者的疗效和安全性。
IBI3016(siRNA,圣因生物/信达生物):目前处于临床2期阶段,1期试验结果在2025年AHA大会上公布,试验结果显示,IBI3016单次给药后,不同剂量组在4周、3个月、6个月时均实现90%左右或以上的AGT水平降低,最大降幅超95%。在3个月时,IBI3016各剂量组均实现血压下降,最高实现24h日间平均动态收缩压/舒张压变化-16.7/-14.7mmHg(安慰剂组为-3.2/-5.7mmHg)、24h夜间动态收缩压/舒张压变化-16.0/-12.9mmHg(安慰剂组为-5.0/-2.6mmHg)。初步结果显示,IBI3016可实现强效且持久的降压效果,且安全性较好。 SAL0132(siRNA,国为生物/信立泰):目前处于临床II期阶段,I期实验结果在2026 ACC公布,在单次剂量递增研究中,所有剂量组均能降低AGT蛋白水平90%以上;潜力治疗剂量组Cohort3在第3个月实现均值约25-13mmHg的诊室血压下降;Cohort4在第2周即观察到DBP下降15.9mmHg,降压效果持续,第6个月仍下降16.8mmHg。
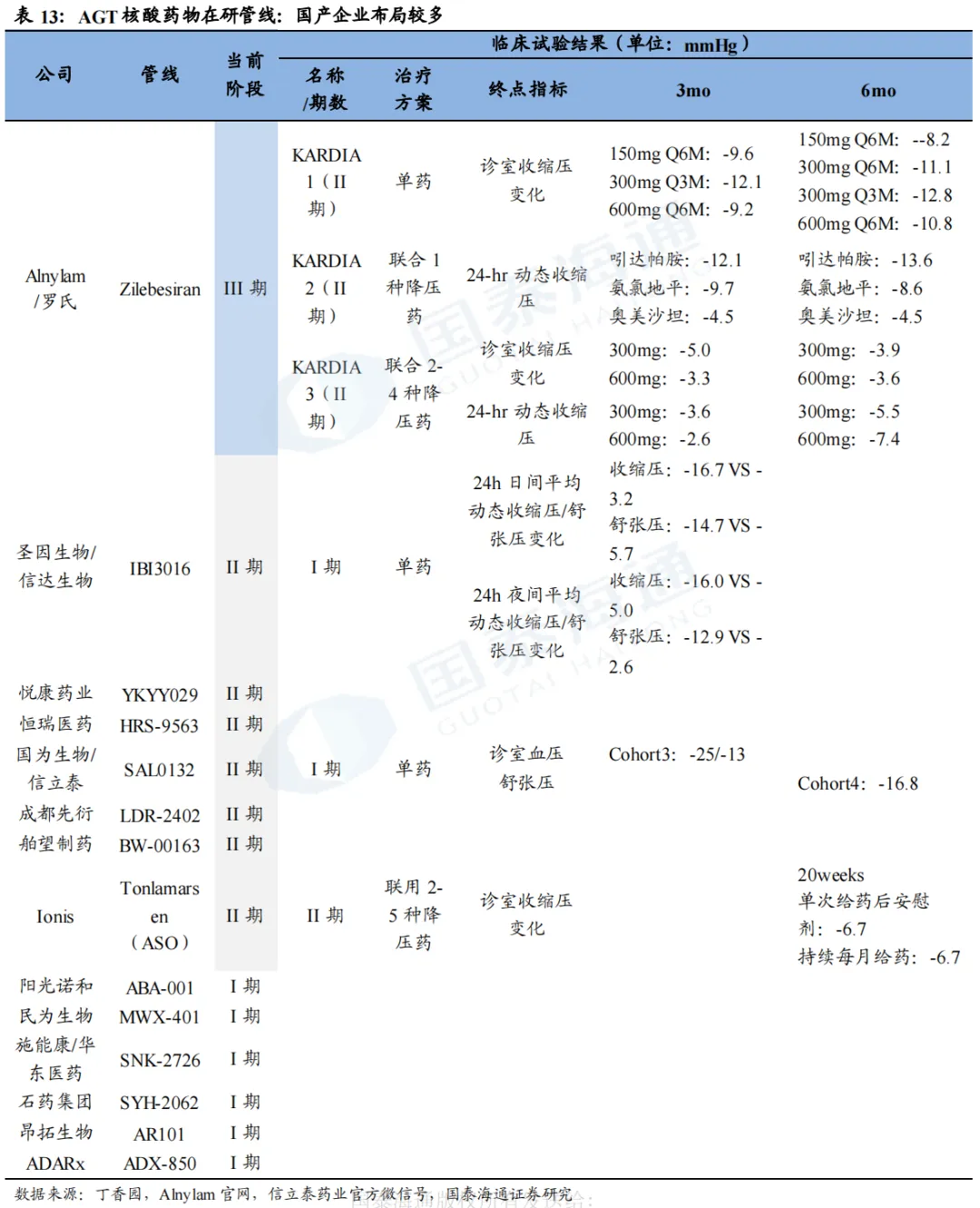
3. 凝血:FXI靶点为蓝海市场,国产领先
3.1. 全球近4000万血栓患者,靶向FXI疗法出血风险低
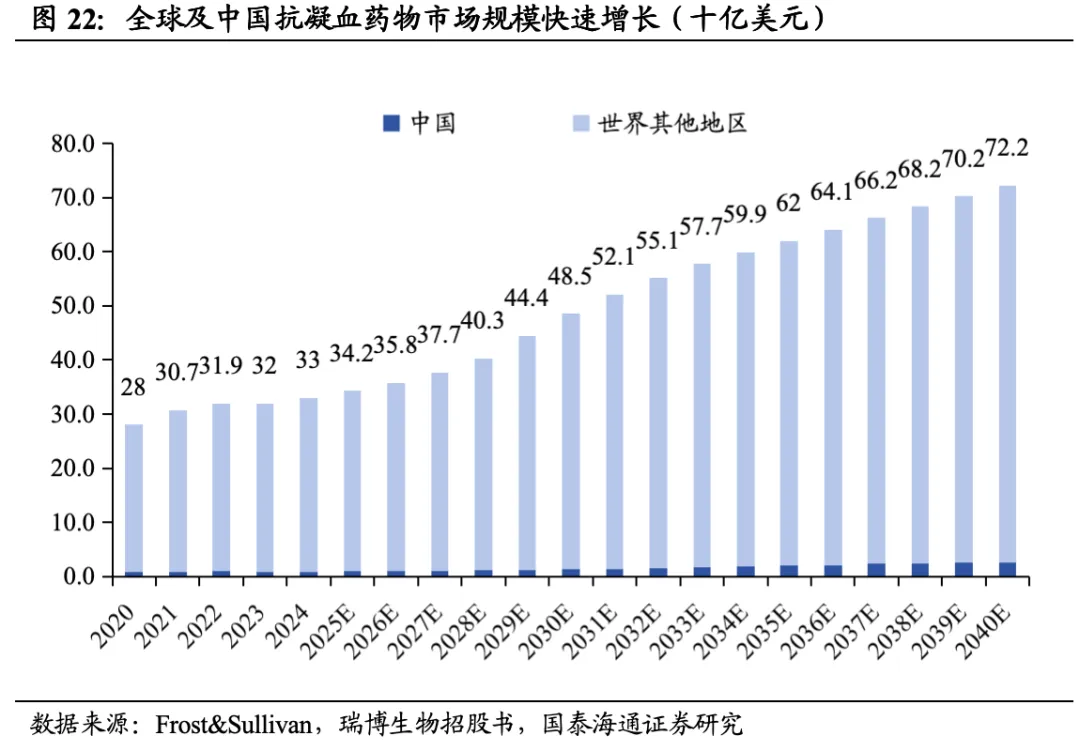
2024年全球近4000万人患有血栓性疾病。血栓性疾病包括一系列以动脉或静脉血管内病理性血凝块形成为特征的疾病。根据瑞博生物招股书,每年全球死亡人数的1/4死于血栓性疾病,抗凝剂市场商业潜力巨大。根据Frost&Sullivan,2024年全球约3860万人患有血栓性疾病,预计2034年达到4160万人。
凝血过程分为内源性通路、外源性通路以及共同通路。内源性通路始于XII因子被激活为XIIa,随后通过一系列级联反应,导致FIXa将FX激活为FXa;外源性通路始于组织因子TF,TF与FVII因子形成复合物,将其激活为FVIIa,随后FVIIa将FX因子激活为FXa;共同通路是内源性通路和外源性通路的结合点,FXa与活化血小板表面的辅因子(FVa因子)结合,形成凝血酶原复合物,该复合物将凝血酶原FII转化为凝血酶FIIa;凝血酶随后将组织蛋白FI裂解为组织蛋白原酶FIa,FIa经聚合形成纤维蛋白凝块。此外,凝血酶将FXIII因子激活为FXIIIa,与纤维蛋白链交联,从而稳定血凝块。
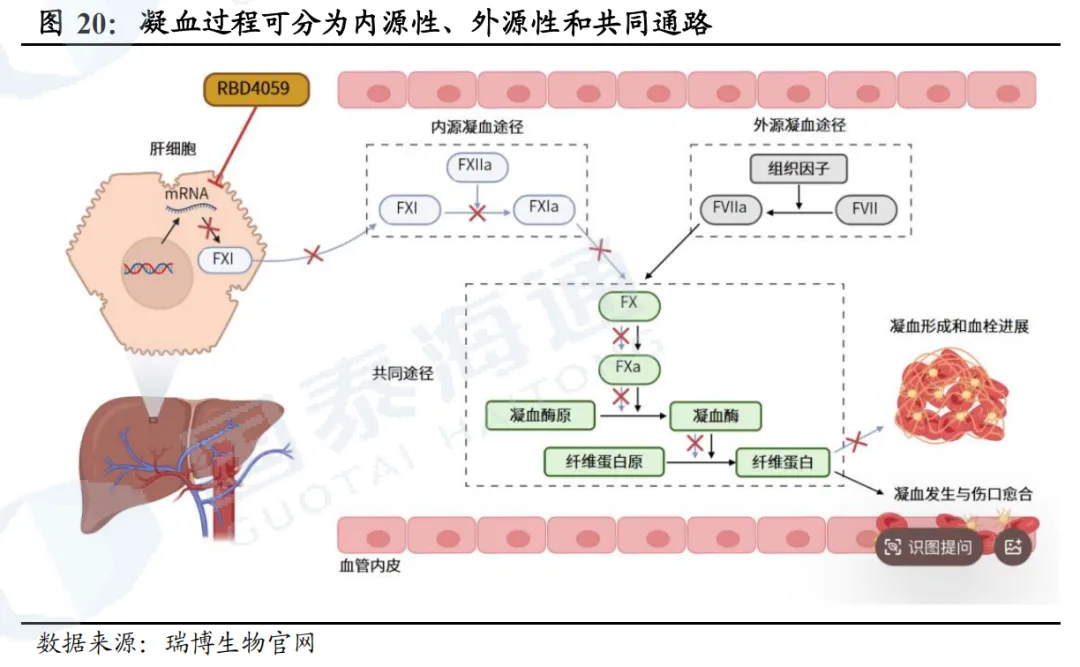
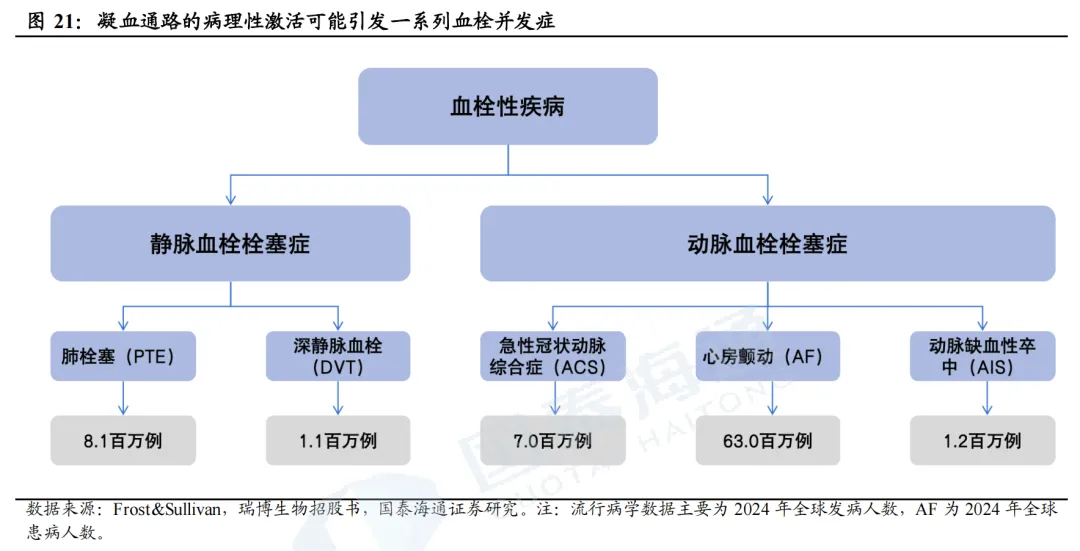
2024年全球抗凝血药物市场规模达330亿美元。目前血栓性疾病的主要药物治疗是抗凝剂,包括华法林、肝素、直接口服抗凝剂(DOAC)等;抗血小板药物主要包括阿司匹林、P2Y12抑制剂等。2024年,全球抗凝血药物市场规模330亿美元,DOAC(直接口服抗凝药)市场规模292亿美元,其中阿哌沙班(Eliquis)销售额207亿美元。在中国,2024年抗凝剂市场规模9亿美元,预计2032年达到15亿美元。
降低出血风险、提高依从性是新型抗凝剂的研发目标。凝血与抗凝之间的平衡受到严格调控,该平衡一旦失调,则可能导致出血性疾病或血栓性疾病。目前已有多种抗凝疗法针对通路的各类因子,如FXa抑制剂(如阿哌沙班)是当前使用最广泛的抗凝疗法,占抗凝剂药物销售总额的约90%。但FXa作用于共同通路,因此会不可避免地损害正常应对损伤的止血反应,从而导致消化道出血、颅内出血等出血并发症的可能性增加。除了出血风险外,依从性挑战(每日用药)、肾功能障碍、药物相互作用等均对FXa抑制剂的使用造成一定挑战。因此,能够有效预防血栓形成、又能最大限度降低出血风险、且依从性佳的药物具有较大的市场需求。
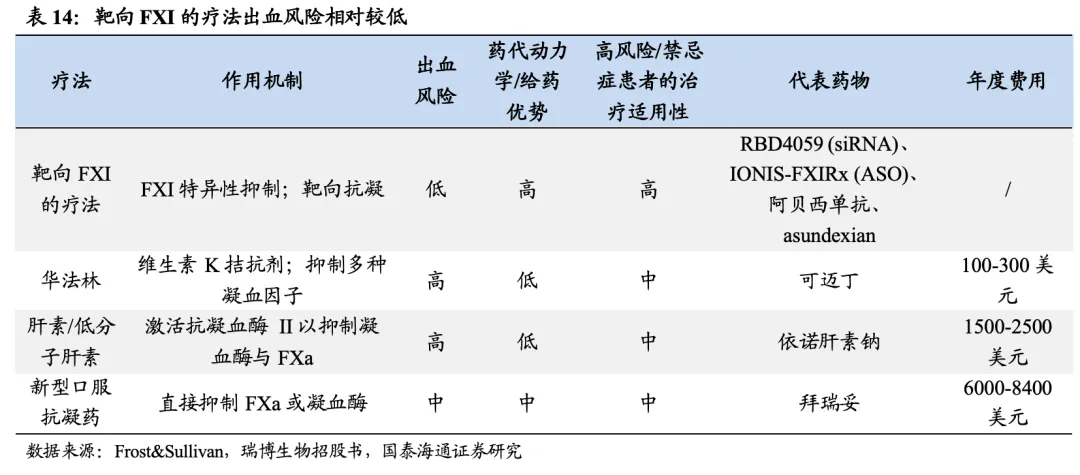
FXI抑制剂为新型抗凝药物,具有安全性和有效性。1)安全性:传统抗凝剂广泛抑制下游通路,而FXI仅对内源性通路进行选择性抑制,保留防止外伤出血所需的生理性止血机制,有望展现出良好的安全性。2)有效性:根据靖因药业招股书,FXI活性低于30%的亚组人群,心血管事件发生率显著降低;FXI低于50%的患者,VTE风险明显降低。
3.2. 竞争格局温和,国产企业全球领先
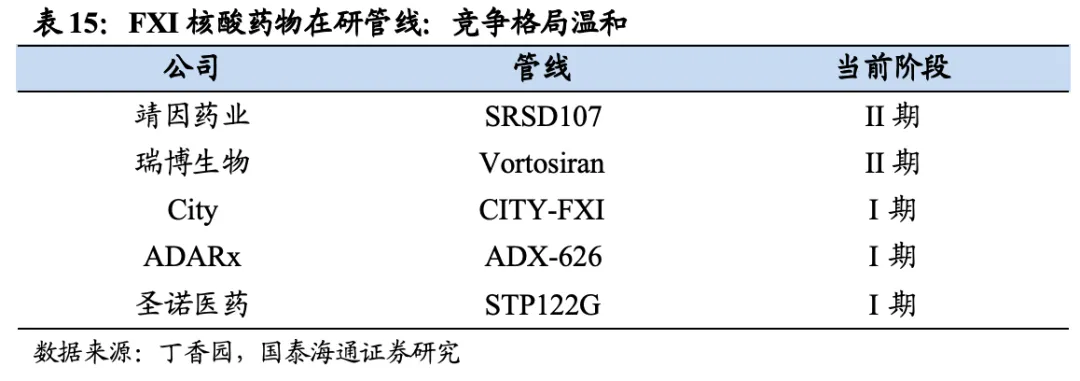
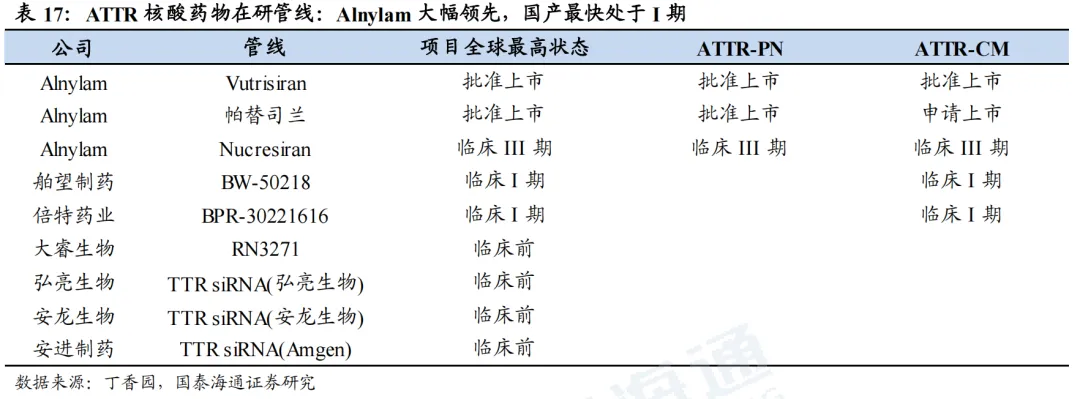
目前,尚无靶向FXI的核酸药物上市,进展较快的为靖因药业的SRSD107、瑞博生物的RBD4059,均处于临床II期阶段,其余药物目前仍处于临床I期或临床前阶段。
Vortosiran/RBD4059(siRNA,瑞博生物)
RBD4059已先后提交2项IIb期临床试验申请。瑞博生物于2024年10月在澳大利亚完成了RBD4059的I期临床试验,并于2024年8月在瑞典启动IIa期临床试验,目前已顺利完成全部患者给药,数据将在2026年举行的心血管学术大会上公布。2026年4月、5月,瑞博生物的RBD4059先后向EMA成功递交针对心房颤动患者卒中预防(SPAF)、静脉血栓栓塞症(VTE)的IIb期临床试验申请(CTA)。此外,瑞博生物的另一款小核酸药物RBD1119也于2026年5月向EMA递交了治疗冠状动脉疾病的II期临床试验申请,持续布局血栓领域。
RBD4059抑制FXI最高超90%,持续性较强。在RBD4059的I期临床试验中,50mg、150mg、400mg、600mg分别实现FXI活性相对基线下降67.5%、81.0%、85.8%、91.6%,疗效可以持续观察到第169天。同时,RBD4059安全性良好,TEAE仅16.7%,且未观察到3级或以上TEAE。抑制水平达到90%以上,也未出现出血风险。
SRSD107(siRNA,靖因药业)
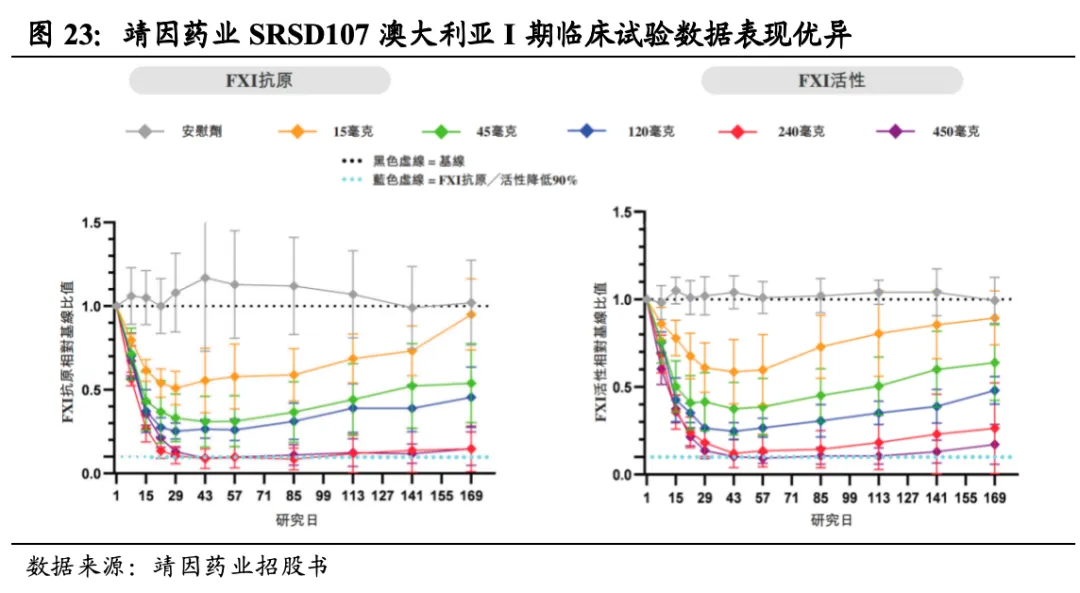
I期实验结果显示SRSD107可降低FXI达95%,作用时间超6个月。靖因药业于2025年5月20日与CRSPER Therapeutics达成合作,以50-50成本和利润分摊机制共同推进SRSD107的开发,靖因药业将获得首付款9500万美元及超8亿美元的预付款和里程碑付款。SRSD107目前已完成两项1期临床试验,结果显示SRSD107可降低循环FXI水平高达95%,作用持续时间超6个月,且活化部分凝血活酶时间(aPTT)相应延长2.4倍,证实了其对内源性通路的有效抑制。单次给药安全性且耐受性良好,除1名接受45mg SRSD107治疗的受试者出现3级严重不良事件(阑尾炎,与治疗无关)外,其余不良事件均为1/2级。
SRSD107的II期临床试验正在推进。靖因药业于2025年9月在欧洲及中国启动SRSD107针对VTE II期多中心临床试验,预计2026H2获得主要疗效终点的初步结果,2027H2完成整体安全性随访;此外,公司于2026年3月已完成SRSD107针对慢性冠状动脉(CAD)和/或外周动脉疾病(PAD)的II期临床试验首例患者给药。
4. 罕见病:以ATTR为锚点,小核酸在罕见病成熟应用
4.1. ATTR全球患者数超35万例,存在较大未被满足需求
ATTR全球患者数量超35万例。ATTR(转甲状腺素蛋白淀粉样变性)核心病因是转甲状腺素蛋白(TTR)错误折叠,形成淀粉样纤维并沉积在全身组织,直接损害器官功能,常见的受累器官包括心脏(引发心肌病,ATTR-CM)和周围神经(引发多发性神经病,ATTR-PN)。遗传型ATTR淀粉样变性(hATTR)全球患者约5万例,主要是TTR基因变异导致蛋白结构异常,属于遗传性罕见病;野生型ATTR淀粉样变性(wtATTR)全球患者超30万例,多与衰老相关(TTR蛋白随年龄增长自发错误折叠)。RNAi疗法可直接靶向肝脏的TTR合成,快速降低TTR水平,阻止淀粉样纤维沉积,从而改善并稳定器官功能,降低死亡/住院风险。
4.2. Alnylam已迭代三代产品,大幅领先其余企业
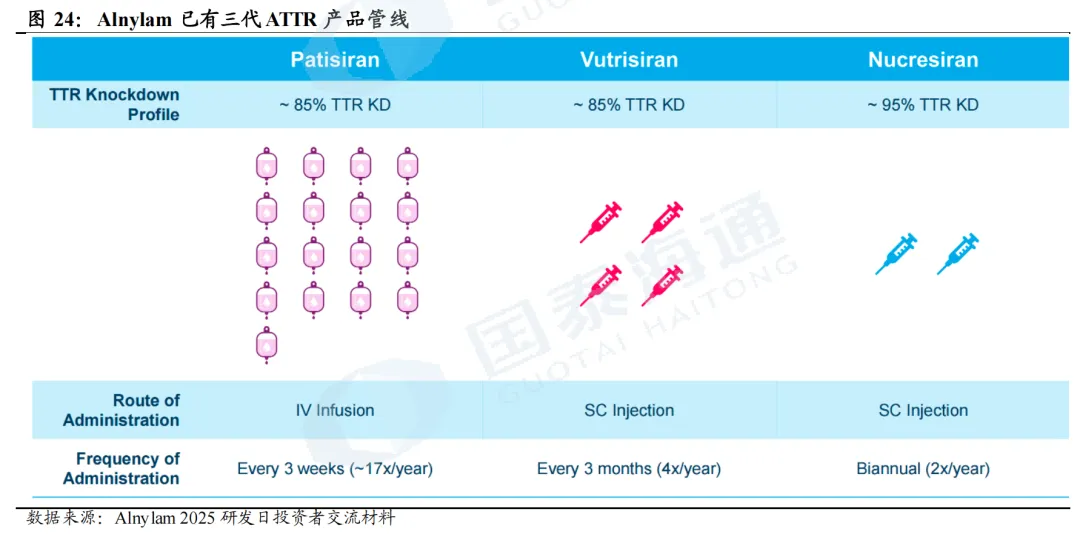
Alnylam是ATTR的代表公司,目前已有三代ATTR产品。其产品迭代历史也彰显了siRNA技术进步的过程,一代产品使用LNP技术;二代产品使用ESC-GalNAc技术,增强其稳定性;三代产品则进一步优化siRNA结构和递送优化目标。由此,产品给药频率逐渐降低,疗效则逐渐升高。具体而言,(1)第一代产品Patisiran(onpattro)仅包含ATTR-PN适应症,于2018-2022年陆续在全球上市,可实现85%的TTR敲低,但给药方式为静脉注射,且每三周一次,便捷性相对较低。(2)第二代产品为Vutrisiran(amvuttra),2022-2024年hATTR-PN适应症陆续上市,2025年ATTR-CM适应症上市,为当前主力产品,可实现85%的TTR敲低,给药方式迭代为每三个月一次皮下给药,大大减少给药频率,增加患者依从性。(3)第三代产品为Nucresiran,目前正处于临床III期阶段,同样为皮下注射,但给药频率有望减少至每年2次,且TTR敲低效率有望提升至95%。根据公司公告,Nucresiran的ATTR-PN适应症有望于2028年读出顶线数据,ATTR-CM适应症则有望于2030年获批上市。
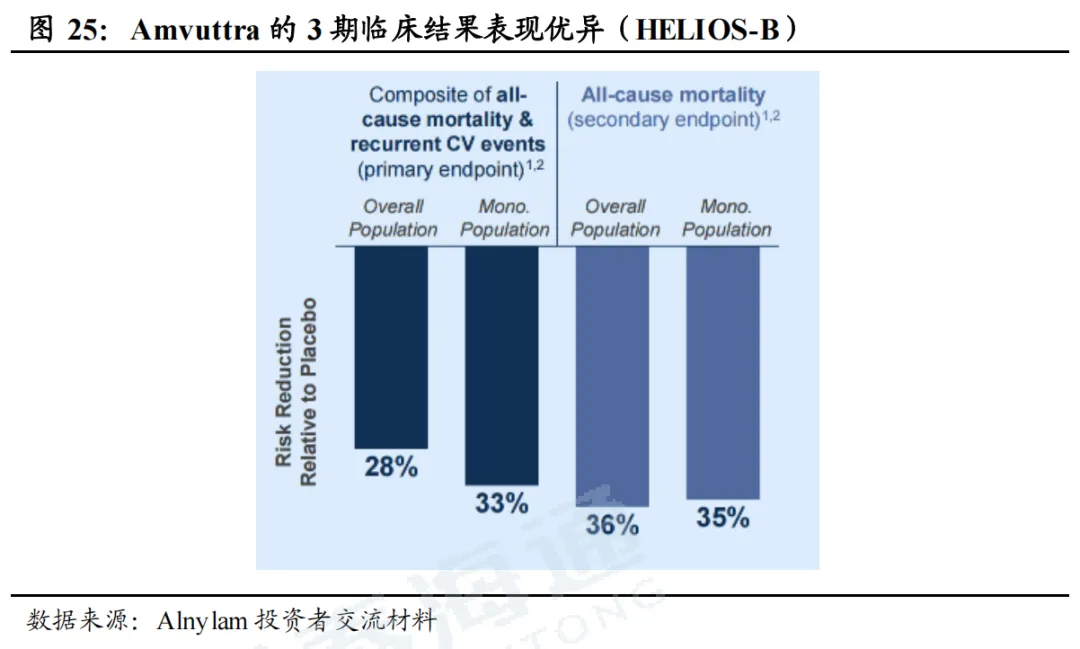
Amvuttra在ATTR-CM适应症中疗效优异。HELIOS-B是一项3期临床研究,用来评估Amvuttra在ATTR-CM患者当中的疗效和安全性,主要终点(复合终点:全因死亡和心血管事件)风险降低28%,单药治疗亚组风险降低33%;次要终点(全因死亡率)风险降低36%,单药治疗亚组风险降低35%。
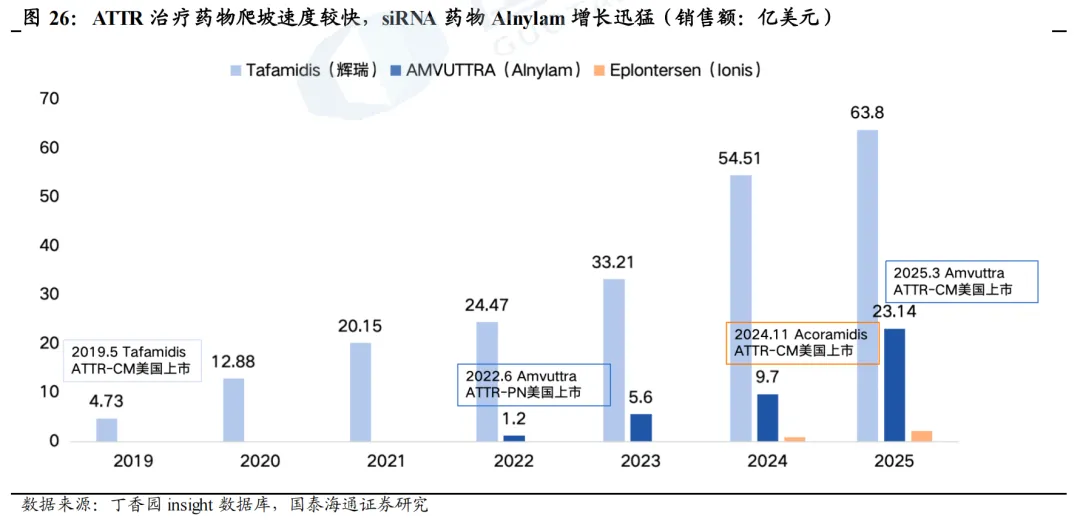
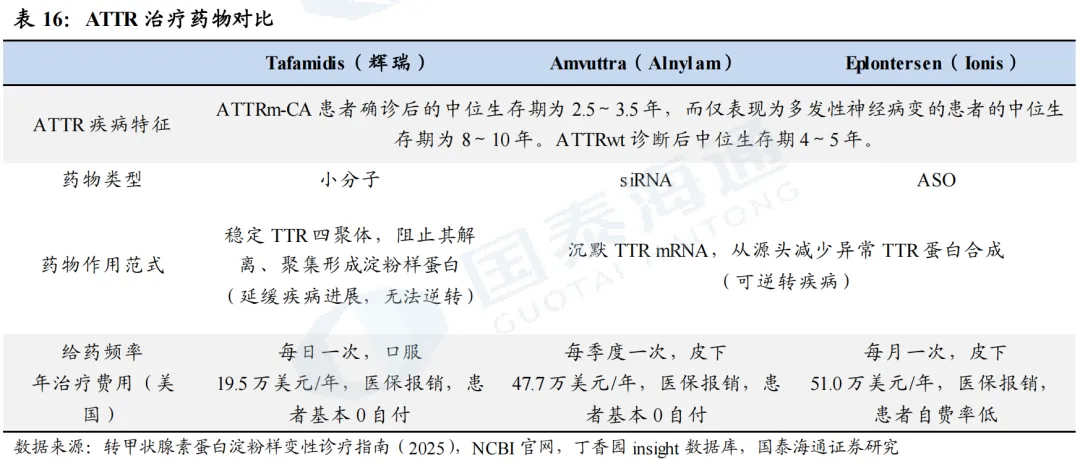
给药频率是销售额拉开差距的重要原因之一。在ATTR-PN适应症方面,Alnylam的Vutrisiran和Ionis的Eplontersen是前后上市的两款靶向TTR的核酸药物,在mNIS+7评分及Norfolk QiL-DN评分方面表现相似,疗效数据同样优异。不同之处在于Vutrisiran为siRNA,给药频率较低(Q3M);Eplontersen为ASO,给药频率相对较高(Q4W)。我们认为给药频率是导致二者上市后销售额相差较大的重要原因之一,2022年6月Vutrisiran在美国获批上市,当年实现1.2亿美元收入,2023、2024年则分别实现5.6亿美元、9.7亿美元收入;而2023年12月Eplontersen在美国获批上市后,2024、2025年仅实现0.85亿美元、2.12亿美元收入。
AMVUTTRA销售额增长迅猛,2025年同比增速达139%。得益于Vutrisiran优异的临床数据和三个月一次的给药频率,以及2025年市场规模更大的ATTR-CM适应症获批,2025年Vutrisiran销售额进一步增加至23.14亿美元,全年销售额同比增长139%,增速迅猛。与之对比,辉瑞的Tafamidis(每日一次,口服)2025年销售额达64亿美元,但增速已回落至17%。
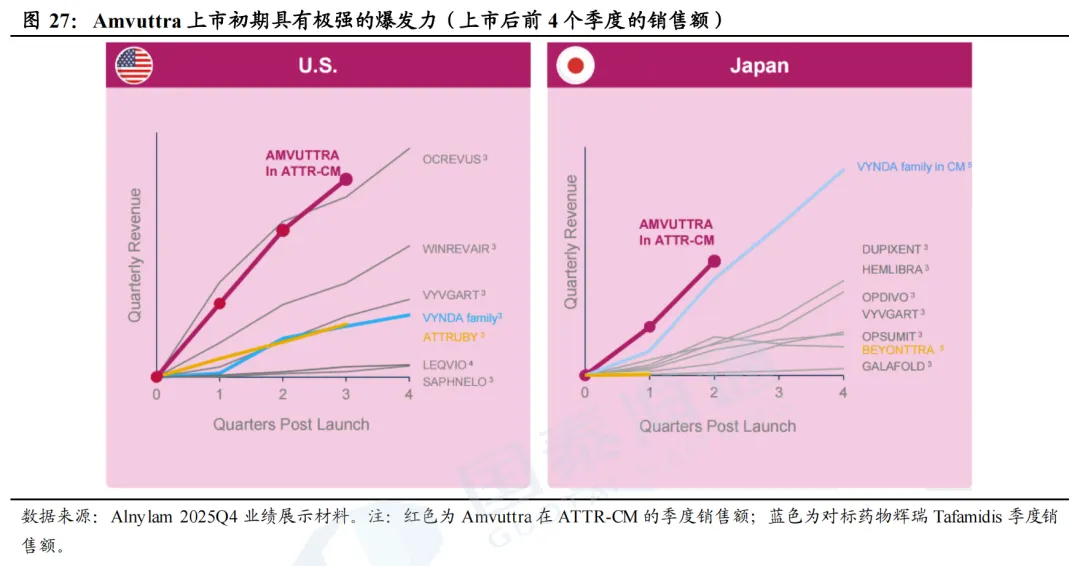
ATTR国内竞争格局较好,仅舶望制药BW-50218和倍特药业BPR-30221616处于临床I期阶段,其余尚处于临床前研究阶段。
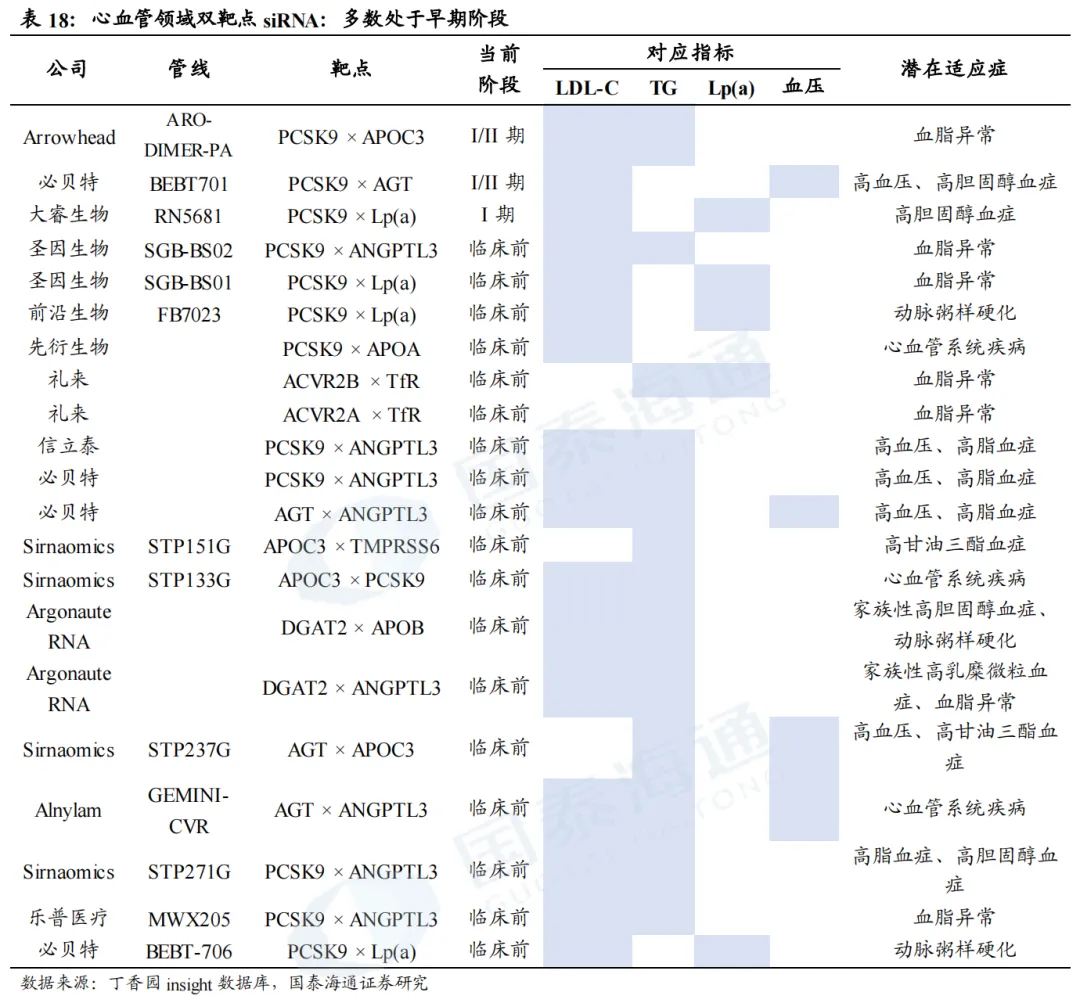
5. 双靶点siRNA:下一个热点研发方向
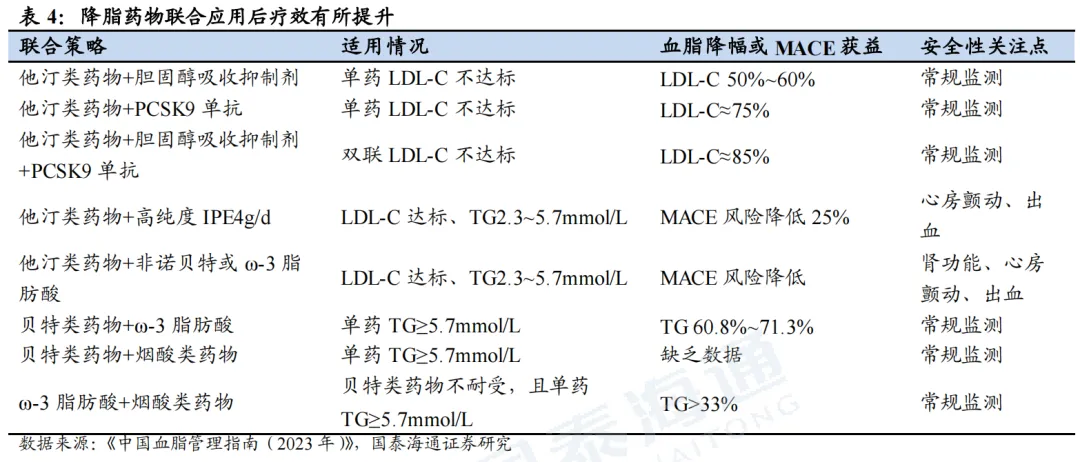
多靶点siRNA是调控复杂多基因异常疾病的潜力方案。许多复杂疾病往往由多基因异常调控诱发,临床治疗需同步靶向多个致病基因。单分子多靶点siRNA可在同一个药物分子上整合多条siRNA序列,实现多基因同步抑制。心血管领域中,各单靶点RNAi降脂药物作用靶点相对单一:PCSK9 siRNA主打降低LDL‑C、APOC3 siRNA以降TG为主、Lp(a) siRNA仅特异性下调Lp(a);ANGPTL3 siRNA虽具备广谱降脂效果,但降脂谱不均衡,降TG作用突出、对LDL‑C降幅有限。而混合型高脂血症患者需要同步管控多项血脂指标,该类患病人群基数庞大,仅美国患者数量便约2000万。另外,血脂异常常诱发高血压、2型糖尿病等代谢合并症,可同步干预多条致病通路的双靶点/多靶点siRNA,是当前临床上尚未被充分满足的重要研发方向。
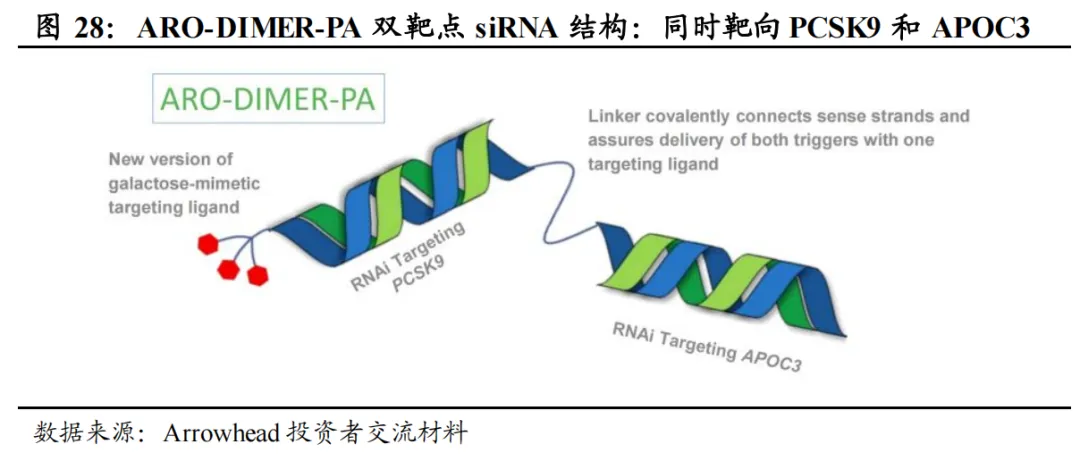
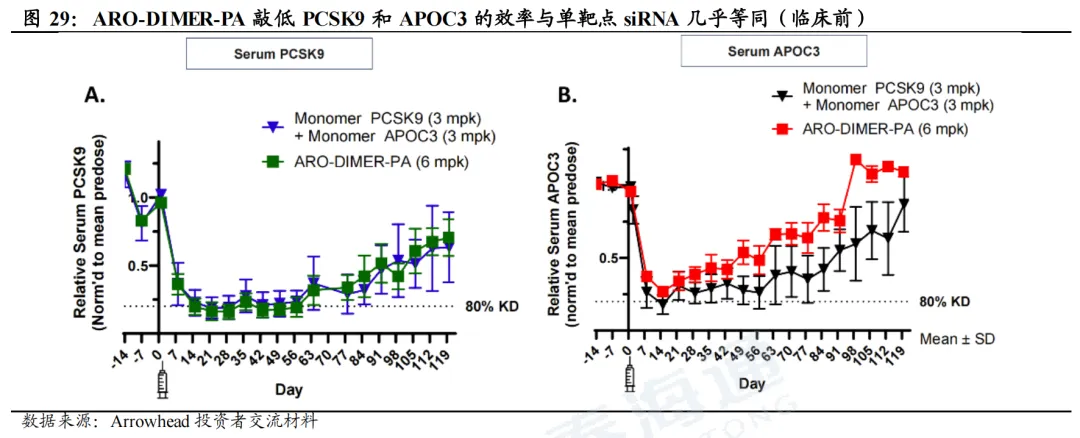
ARO-DIMER-PA(Arrowhead):全球进度最靠前的双靶点siRNA,预计2026Q3读出临床I/II期数据。ARO-DIMER-PA可同时靶向PCSK9和APOC3的siRNA,从而有望同时降低LDL-C及TG的水平。临床前(食蟹猴)实验结果表明,1)ARO-DIMER-PA具有靶向优势,给药后>30%的药物富集肝脏;2)ARO-DIMER-PA(6mg/kg)给药与PCSK9 siRNA(3mg/kg)+APOC3 siRNA(3mg/kg)效果类似;3)单次皮下注射ARO-DIMER-PA后,LDL-C、非HDL-C、TG水平同步下降近50%。
BEBT-701(必贝特):2026年2月,BEBT-701获得NMPA批准临床,开展轻中度高血压合并低密度脂蛋白胆固醇升高的I/II期临床试验。BEBT-701是依托必贝特专有的GalNAc双寡核苷酸偶联(GDOC)技术平台开发的首个进入人体研究的新药,可通过单一分子同时沉默AGT和PCSK9。非临床研究结果显示,BEBT-701在人源化小鼠模型与非人灵长类动物中对AGT与PCSK9均实现高效且持久的基因沉默,在降压与降脂方面表现出协同药效优势,并具备良好的安全性特征。
2MW7141(迈威生物/Kalexo):2025年9月,迈威生物将2MW7141全球范围的独家权利授权给Kalexo,获得10亿美元预付款和里程碑付款,以及低个位数的特许权使用费,且获得Kalexo总计双位数的A轮优先股。2MW7141是一款处于临床前的双靶点siRNA,主要针对血脂异常人群的血脂调控以及高危心血管事件的预防。
6. 风险提示
技术转化不及预期,后续临床试验失败风险。

如需获取全文,请联系国泰海通政策和产业研究院、国泰海通对口机构销售。

国泰海通政策和产业研究院

国泰海通政策和产业研究院立足国家战略全局与全球竞争格局,对外致力于打造具有行业引领力与重要影响力的高端智库,面向政府、企业与机构投资者,提供前瞻性政策研判、深度产业洞察与国别研究支撑,服务国家治理现代化与产业体系升级,支撑高质量发展与高水平对外开放;对内强化“投研—投资—投行”协同联动,强化研究引领与业务赋能,推动研究成果向公司综合服务能力与协同优势高效转化,巩固拓宽公司市场竞争护城河。
重要提醒
本订阅号所载内容仅面向国泰海通证券研究服务签约客户。因本资料暂时无法设置访问限制,根据《证券期货投资者适当性管理办法》的要求,若您并非国泰海通证券研究服务签约客户,为保证服务质量、控制投资风险,还请取消关注,请勿订阅、接收或使用本订阅号中的任何信息。我们对由此给您造成的不便表示诚挚歉意,非常感谢您的理解与配合!如有任何疑问,敬请按照文末联系方式与我们联系。
法律声明
本公众订阅号为国泰海通证券股份有限公司(以下简称“国泰海通证券”) 政策和产业研究院依法设立、独立运营的唯一官方订阅号。其他机构或个人在微信平台上以国泰海通证券政策和产业研究院名义注册的,或含有与国泰海通证券政策和产业研究院品牌名称相关信息的其他订阅号均不是国泰海通证券政策和产业研究院官方订阅号。
本订阅号不是国泰海通证券研究报告发布平台,本订阅号所载内容均来自于国泰海通证券政策和产业研究院已正式发布的研究报告,如需了解详细的证券研究信息,请具体参见国泰海通证券政策和产业研究院发布的完整报告。
在任何情况下,本订阅号的内容不构成对任何人的投资建议,国泰海通证券也不对任何人因使用本订阅号所载任何内容所引致的任何损失负任何责任。
本订阅号所载内容版权仅为国泰海通证券所有,国泰海通证券对本订阅号保留一切法律权利。订阅人对本订阅号发布的所有内容(包括文字、影像等)进行复制、转载的,需注明出处为“国泰海通研究”, 且不得对本订阅号所载内容进行任何有悖原意的引用、删节和修改。